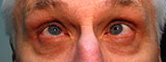
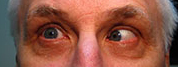
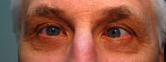
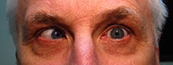
History of Present Illness: A 46-year-old male, presented for follow up of a long-standing progressive esotropia (ET). In 1986, he presented with 14 prism diopters of ET and was noted to have bilateral abduction deficits.
In 1994, he was seen by a neuro-ophthalmologist. It was noted that he was wearing 3 prism diopters of base out prism in his glasses. He had 36 prism diopters of ET, but no diplopia. Worth 4 dot testing showed that he was suppressing his left eye. He continued to have bilateral abduction deficits now with slow saccades of the lateral recti and end gaze nystagmus with lateral gaze. The patient brought in old photos at a follow-up exam which showed no ET as a child.
He had been followed for several years for long-standing ET, but was never given a firm diagnosis. At his last visit in 2008, he had stopped wearing prisms because they were of no help to him. He still had no diplopia.
PMH/FH/POH: Patient carried a diagnosis of primary progressive cerebellar ataxia. He reported being very clumsy as a teenager. In 1972, he developed ataxia. In 1989, an MRI of the brain revealed cerebellar and medullary atrophy. Based on this MRI and lack of family history, he was given a diagnosis of primary progressive cerebellar ataxia. He knows very little of his parent's medical history and has no siblings or children.
OCULAR EXAM:
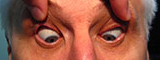
Figure 1-5: Patient had 52 prism diopters of ET in primary gaze with bilateral abduction deficits.
|
|
|
|
|
|
|
|
|
Video 1: Extraocular movements. Note the bilateral abduction deficits and end gaze nystagmus on lateral gaze. |
|
NEUROLOGIC EXAM:
Video 2: Dysdiadokinesis with finger-to-nose testing |
|
Video 3: Ataxic gait |
|
Video 4: Dysarthria |
|
This gentleman originally presented in 1986 with progressive esotropia. Since that time, there have been multiple publications linking spinocerebellar ataxia to disorders of extraocular movements and vergence. This gentleman was relieved to hear that his long-standing esotropia was likely related to his spinocerebellar ataxia. Despite his negative family history, it is possible that one of his parents could have carried a milder form of the disease due to anticipation.(see below for explanation).
It was discussed with him that he could undergo strabismus surgery if he liked, however his esotropia would likely continue to progress. This would likely cause diplopia, which he currently did not have secondary to ignoring. He was seen one year later in follow up after this exam. His ET was stable.
Diagnosis: Spinocerebellar ataxia with ophthalmoplegia
There are at least 30 different types of spinocerebellar ataxias (SCA) also referred to as autosomal dominant cerebellar ataxias (ADCA). These are divided into three categories based on they're typical presentation (See Table 2 below). The number of ADCAs increases each year as more protein products are genotyped (1).
Several of the SCAs have known protein products such as ataxin-1 which is responsible for SCA 1. Most SCA mutations involve CAG nucleotide repeats resulting in protein products with large polyglutamine domains. These proteins tend to aggregate and form inclusions. These inclusions have a toxic effect. CAG repeats are unstable leading to de novo mutations that can occur during transmission and cause trinucleotide repeat expansion. This leads to anticipation meaning that the phenotypical expression can increase between generations (2,3). It is important to remember when patients present with apparent sporadic disease, their parents may have had a less severe form of the disease.
The most prevalent SCA worldwide is SCA3 also known as Machado-Joseph Disease. This disease was named for two families of Portuguese and Azorean decent described in the 1970s. The prevalence is 1 in 4000 among Portuguese and 1 in 140 on the Azorean island of Flores. It has been associated with a higher incidence of diplopia and ophthalmoplegia than other SCAs (4).
Cerebellar disease can cause multiple disorders of ocular motility. Recently there has been clear evidence of impairment of vergence eye movements in SCA3. The anatomical centers that mediate vergence movements are still not completely understood. There is some evidence that the medial nucleus reticularis tegmenti pontis (NRTP) may be involved. Interestingly, the NRTP is contiguous with the paramedian pontine reticular formation (PPRF) which controls lateral gaze (4).
Slow saccades SCA1, SCA2, SCA3, SCA7, SCA28 Down-beat nystagmus SCA6 Ophthalmoplegia SCA1, SCA2, SCA3, SCA28, SCA30 Ocular dyskinesia SCA10 |
Aside from disorders of ocular motility, a characteristic finding of SCA7 is cone-rod dystrophy. Recent evidence suggests that ataxin-7 interacts with CRX which is a nuclear transcription factor predominantly expressed in retinal photoreceptor cells in which a mutation can cause cone-rod dystrophy.(5)
EpidemiologyPrevalence 1-4 of 400,000 Age of onset varies based on mutation but onset is typically 3rd-4th decade Spinocerebellar Ataxia implies an autosomal dominant pattern. There are several other inherited, sporadic, and acquired causes of cerebellar ataxia (1).
|
Signs
|
SymptomsImbalance with standing Impaired coordination Slurred speech Diplopia |
TreatmentLimited Some results with 5-hydroxytryptophan, buspirone or tandospirone, sulfamethoxazole/trimethoprim or lamotrigine in SCA3 Strabismus surgery if stable (6) |
Privett BK, Kardon RH. Spinocerebellar Ataxia with Ophthalmoplegia: 46-year-old male presenting with progressive esotropia. EyeRounds.org. February 16, 2010; Available from: http://www.EyeRounds.org/cases/109-SCA.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links