
Chief Complaint: Decreased vision and glare in both eyes.
History of Present Illness: A 28-year-old female with a history of Marfan syndrome presented to the comprehensive ophthalmology clinic reporting a progressive decrease in vision and worsening glare in both eyes. She had been seen by ophthalmologists in the past, and had been told that her crystalline lenses were subluxed in both eyes. She had not had problems with her vision until recent months.
Medical History: Marfan syndrome with aortic stenosis followed by cardiology
Medications: Oral beta blocker
Family History: No known family members with Marfan Syndrome. Grandmother with glaucoma
Social History: The patient is a graduate student.
 |
 |
The patient’s subluxed lenses led to poor vision from peripheral lenticular irregular astigmatism and glare. She was taken to the operating room where her relatively clear lenses were removed and iris sutured intraocular lenses were placed. The surgical video for one eye is shown below.
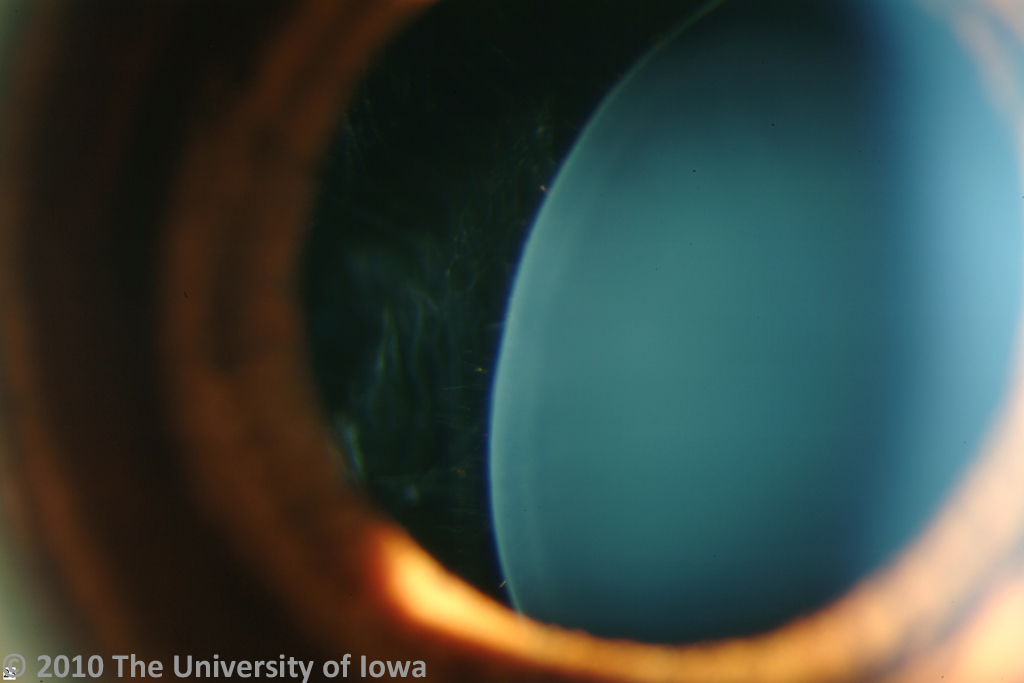
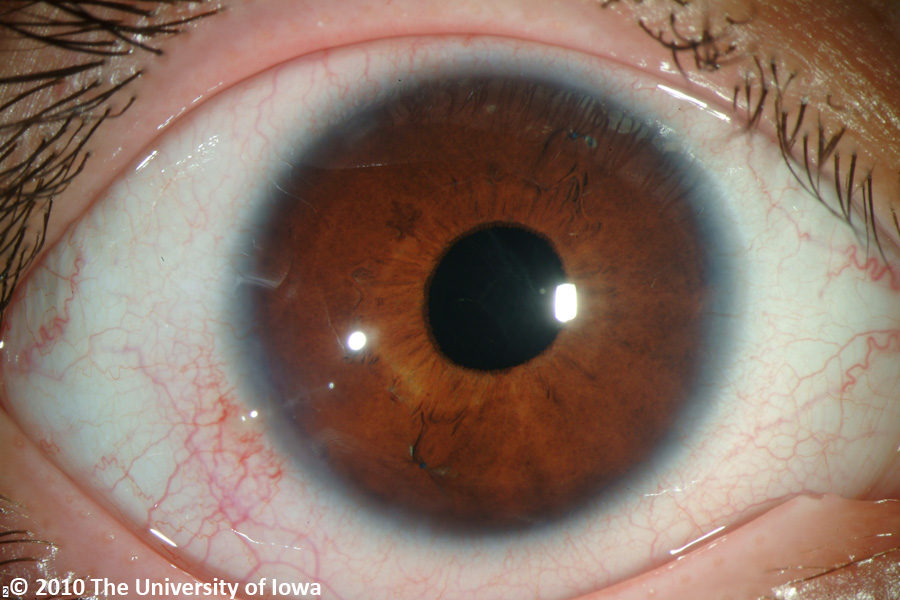
Her post operative course was unremarkable with excellent improvement in her visual function. Her iris-sutured intraocular lenses have remained stable for several years as shown in figure 3 and 4.
 |
 |
Marfan syndrome is a pleotropic autosomal dominant genetic disorder that results in weakening of connective tissue in the musculoskeletal, cardiovascular and ocular organ systems. It is the second most common inherited connective tissue disorder, with an incidence of between 1/5,000 and 1/20,000. The abnormality in over 80% of Marfan patients involves defects in the protein fibrillin 1 (FBN1) on chromosome 15, a structural component of microfibrils found in connective tissue throughout the body. More than 500 different mutations in the FBN1 gene have been indentified. Another mutation believed to result in the Marfan phenotype is the inactivation of the TGF-β receptor 2 (TGFBR2), thought to disrupt the integration of fibrillin into connective tissue.
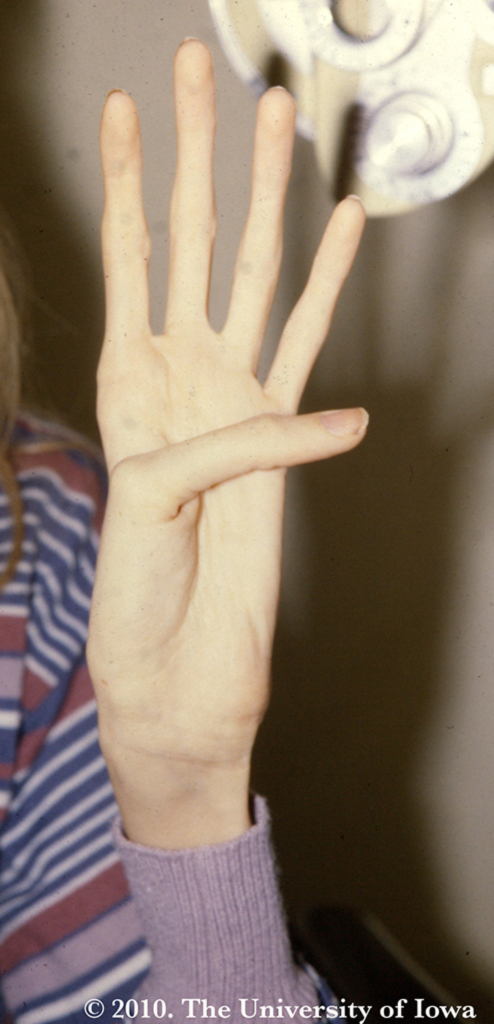 |
The diagnosis of Marfan syndrome is complicated by a genotype-phenotype inconsistency that presents with widely variable clinical manifestations. Clinical diagnosis is currently based on Ghent Nosology (1996) which specifies the involvement of at least 3 organ systems (Skeletal, ocular, cardiovascular, dura mater, pulmonary, or skin/integument), 2 of which must meet "major criteria." Marfan syndrome can also be diagnosed with equivocal genetic testing, which can be offered to patients in the context of family planning and pedigree formation.
The systemic manifestations of Marfan syndrome are well-studied, the most obvious of which are the musculoskeletal abnormalities. Again, the common thread in the variety of Marfan phenotypes is the weakness or incompetence of the connective tissues due to defects in fibrillin. The Marfan patient is often very tall with long, flexible extremities and marked scoliosis. They exhibit arachnodactyly (spider fingers, see Figure 5) with the ability to dramatically encircle the wrist (Walker-Murdoch sign). In addition, they often have pectus excavatum, a high-arched palate, and facial abnormalities. The cardiovascular findings range from mild mitral valve prolapse to severe aortic aneurysm or dissection; the severe cardiovascular complications are the primary causes of mortality among Marfan patients. Pulmonary diseases include apical blebs or spontaneous pneumothorax.
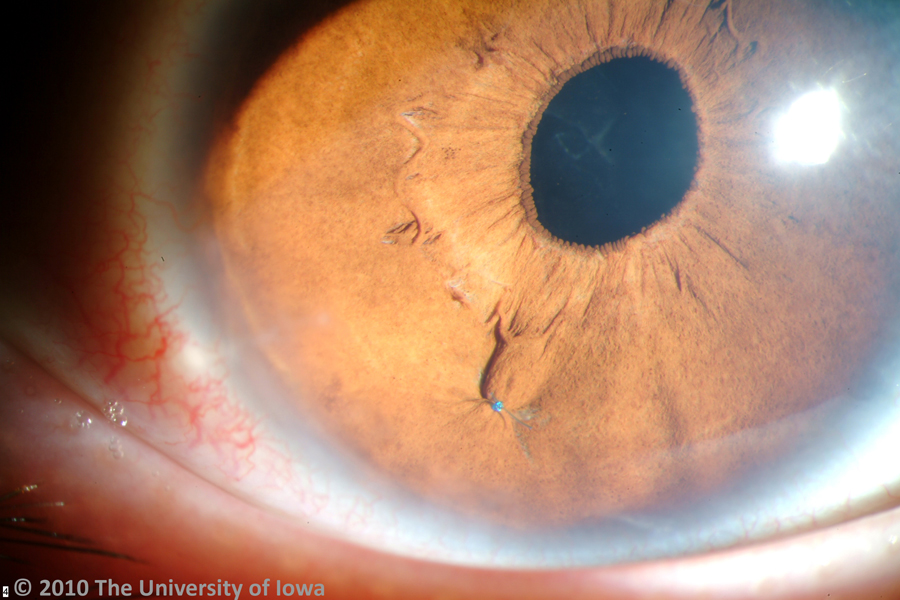
The major ocular abnormality in Marfan syndrome is ectopia lentis (lens subluxation or dislocation, see Figure 6). While relatively little is known about the exact mechanism of this ocular pathology in Marfan syndrome, a number of theories have been suggested. Wheatley et. al (1995) found that while fibrillin is localized to the superficial capsule and ciliary epithelial surface at the attachment of the zonules in normal eyes, Marfan patients lack such localization and exhibit abnormal ciliary processes with absent or severely disorganized zonules. This pathology was found to be positively correlated with lens subluxation. Clinically, ectopia lentis is bilateral in 60-87% of Marfan patients and is stable from childhood. Symptoms include fluctuating blurred vision, monocular diplopia, and pain. On exam, patients demonstrate refractive instability with myopia and astigmatism, iridodonesis, phacodinesis, and a recessed angle. The most common direction of dislocation on exam is superotemporal. In addition, there may be secondary complications from lens movement such as phacolytic uveitis from posterior subluxation of the lens to the vitreous.
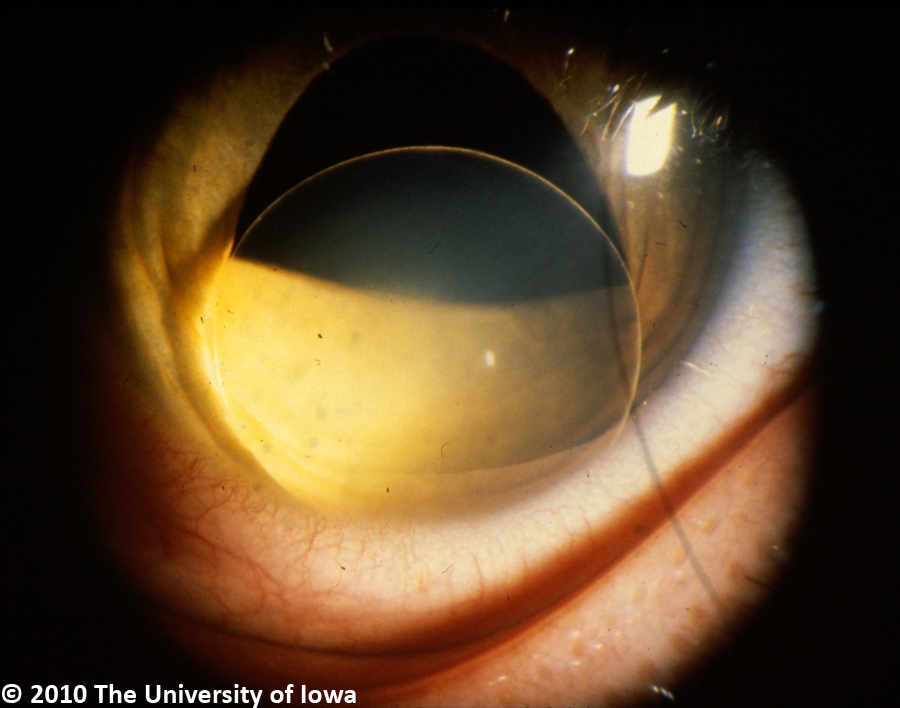 |
Retinal tears and detachments are also quite common in patients with Marfan syndrome. The Marfan eye has many risk factors for tears and detachments including high myopia, excess lattice degeneration, vitreous liquefaction, choroidal and scleral thinning, and vitreous traction from ectopia lentis. Each of these changes is thought to be related to fibrillin alterations. Retinal detachment occurs in 5-26.5% of patients with Marfan syndrome and is bilateral in 30-42% of these cases. Rigorous screening and close follow-up of symptoms is important to prevent retinal detachment in these cases.
Other ocular manifestations of Marfan syndrome include flattened cornea (causing astigmatism), keratoconus, increased globe length (causing myopia), iris coloboma, cataracts, glaucoma, strabismus, amblyopia, and vascular malformations. Central to each of these findings is the ubiquitous abnormality of fibrillin in the ocular connective tissue of the Marfan patient.
With regard to ectopia lentis, a number of treatment options exist. Mild subluxation allows for near normal vision with the patient seeing through the phakic portion of the pupil. The other extreme would be severe subluxation in a child which requires urgent surgical correction to avoid irreversible amblyopia. The principle surgical method employed in Marfan syndrome is lens extraction with either IOL placement or contact lens correction. Surgery is indicated when the lens position causes irregular astigmatism and glare, when the lens is posteriorly dislocated into the vitreous, when the lens is dislocated anteriorly and causes secondary glaucoma, or in the setting of cataract formation. Because of the altered anatomy and weakening of the connective tissues in Marfan syndrome, all of the usual complications of lens extraction are amplified. As the capsule is unstable from loss of zonules, the intraocular lens (IOL) must either be placed in the anterior chamber, or be secured to the iris or sclera with permanent prolene sutures.
One important complication of IOL fixation to the sclera in young patients is that the 10-0 prolene sutures may break three to ten years following surgery. This has led many surgeons to use larger suture such as 8-O gortex or 9-O prolene in the hopes that the suture will last longer. In cases of complete posterior lens dislocation, a pars plana vitreolensectomy can be performed. Another surgical option is to secure the capsule to the sclera using sutured cionni modified capsular tension rings (CTR from Morcher) or Ahmed capsular tension segments (CTS from Morcher). These surgical interventions, while complicated, have great success in restoring vision in these patients.
EPIDEMIOLOGY
|
SIGNS (Ocular)
|
SYMPTOMS
|
TREATMENT of ocular complications
|
Welder J, Nylen EL, Oetting TA. Marfan Syndrome. EyeRounds.org. May 6, 2010; Available from: http://www.EyeRounds.org/cases/110-Marfan.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
