
A 73-year-old woman with pseudophakia returns for one-year follow-up.
Since cataract extraction last year, the patient has enjoyed her improved vision with no complaints.
Pseudophakia, both eyes, 2009
Posterior YAG capsulotomy, left eye
Refractive error and presbyopia, both eyes
Obesity, hypertension, hypercholesterolemia
No ocular medications. Systemic medications: amlodipine, atorvastatin, hydrochlorothiazide
No family history of glaucoma, macular degeneration, blindness or known ocular diseases.
Smoking history of 17 pack years, quit in 1975. Alcohol use is <1 drink per week.
Visual acuity (distance with correction):
Right eye (OD): 20/30, pinhole to 20/25
Left eye (OS): 20/40, pinhole to 20/25
Intraocular pressure (applanation): OD 13, OS 14
Pupils: Equal, round, 3 mm in dark, 2 mm in light, no relative afferent pupillary defect
Visual fields: Full to confrontation both eyes (OU)
Motility: Full OU
 |
 |
Based on the clinical appearance and asymptomatic nature of her corneal opacities, the patient was diagnosed with polymorphic amyloid degeneration.
Since visual acuity was not affected, no treatment was necessary.
The patient continued yearly routine exams and her vision remained stable, with only small changes in refractive error. Her exam also remained stable with no change in the quantity or quality of the corneal opacities.
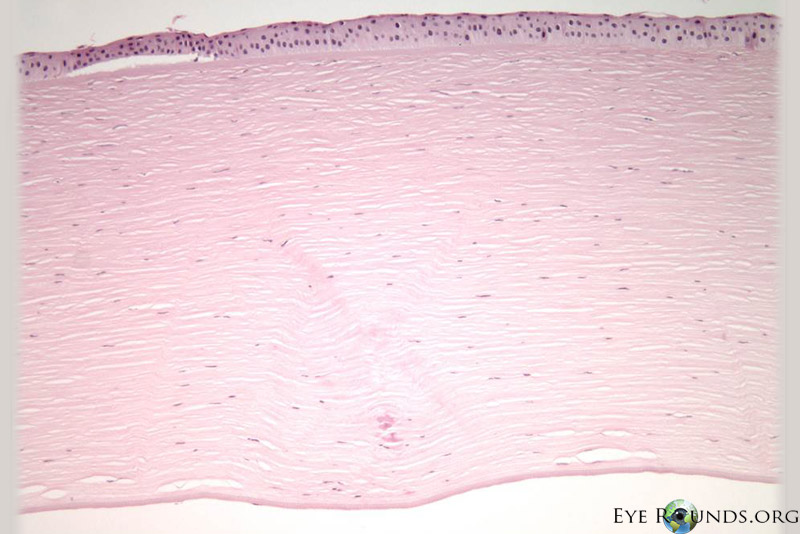
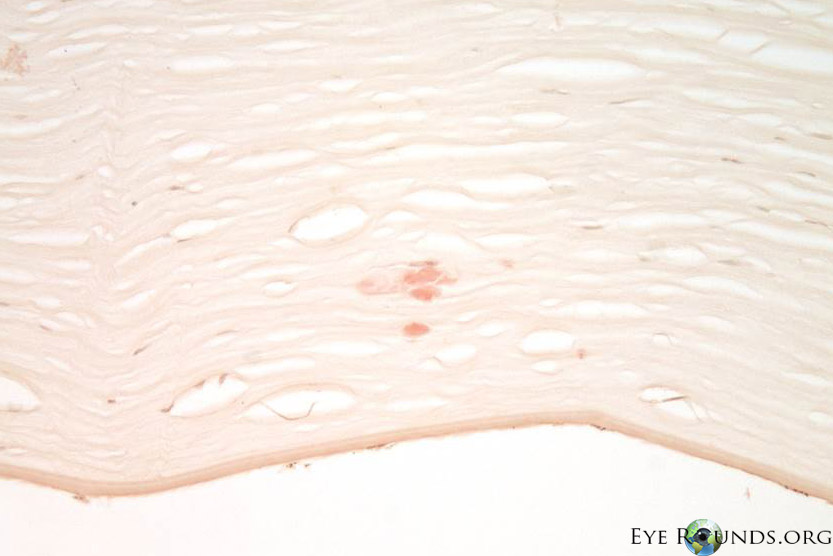
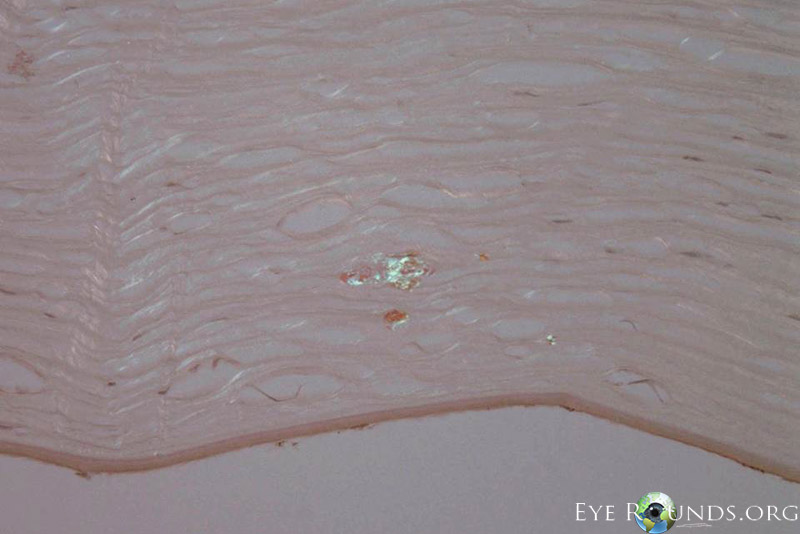
Polymorphic amyloid degeneration is not visually significant, and corneal transplantation is not indicated. However, when intrastromal amyloid is identified incidentally in corneal tissue obtained for other reasons, polymorphic amyloid degeneration is on the differential diagnosis. There are no histology slides from this patient’s case because polymorphic amyloid degeneration does not cause decreased visual acuity. Figure 2 A,B,C shows the histopathology findings of polymorphic amyloid degeneration in a different patient who had a penetrating keratoplasty for pseudophakic corneal edema.
 |
 |
 |
Polymorphic amyloid degeneration is a bilateral non-heritable condition of middle and old age characterized by non-progressive amyloid deposition within the mid and deep corneal stroma. The deposits form polymorphous and filamentous opacities in an axial distribution [1]. There is no associated inflammation, vascularization, or altered visual acuity [2].
There has been no identified genetic basis of inheritance of polymorphic amyloid degeneration [3,4]. Onset of disease is in people ages 50 years or older, and the amyloid deposits do not appear to progress [3]. Polymorphic amyloid degeneration is thought to be sporadic, with no clear sex predominance, nor any consistent association with systemic or ocular disease, although it has been identified in case reports of unrelated diseases [1,5,6].
The diagnosis is made by slit lamp examination, which reveals gray-white axial deep stromal opacities of the cornea. Clear, refractile, stromal filamentous opacities are evident by retroillumination [7].
Histopathological diagnosis, which is not required and only found incidentally on patients that undergo corneal transplantation for other reasons, may be made with Congo red staining of the amyloid in the deep stroma, with dichromaticism under polarized light. Electron microscopy shows filaments and punctate lesions woven between normal stromal collagen [5].
The name polymorphic amyloid degeneration was suggested by Mannis et al in 1975. The previous name, polymorphous stromal dystrophy, was suggested by Thomsitt and Bron [2]. The reason this condition was renamed as a degeneration instead of a dystrophy was the lack of evidence of heritability, and its detectable presence only in people greater than 50 years of age.
Since the diagnosis of polymorphic amyloid degeneration is a clinical diagnosis, an additional example of polymorphic amyloid degeneration is displayed in Figure 3.
 |
For a summary of the key facts regarding polymorphic amyloid degeneration, see Table 1 below.
Table 1
Epidemiology
|
Signs
Diagnosis
|
Symptoms
|
Treatment
|
Polymorphic amyloid degeneration is a condition characterized by stromal gray-white opacities that do not progress or impair visual acuity after presentation in the sixth decade of life. The diagnosis is based on clinical examination. Any change in vision should prompt a search for other causes or reconsideration of the diagnosis of polymorphic amyloid degeneration.
1. Mannis MJ, Krachmer JH, Rodrigues MM, Pardos GJ. Polymorphic amyloid degeneration of the cornea. A clinical and histopathologic study. Arch Ophthalmol. 1981; 99:1217-1223. [PMID: 6973332]
2. Thomsitt L, Bron AJ. Polymorphic stromal dystrophy. Br J Ophthalmol. 1975;59:125-132. [PMID: 1079457]
3. Molia LM, Lanier JD, Font RL. Posterior polymorphous dystrophy associated with posterior amyloid degeneration of the cornea. Am J Ophthalmol. 1999 Jan;127(1):86-8. [PMID: 9933006]
4. Nirankari VS, Rodrigues MM, Rajagopalan S, Brown D. Polymorphic amyloid degeneration. Arch Ophthalmol. 1989 Apr;107(4):598. [PMID: 2705931]
5. Krachmer JH, Dubord PJ, Rodrigues MM, et al. Corneal posterior crocodile shagreen and polymorphic amyloid degeneration. Arch Ophthalmol. 1983; 101:54-59. [PMID: 6600392]
6. Waring GO 3rd, Malaty A, Grossniklaus H, Kaj H. Climatic proteoglycan stromal keratopathy, a new corneal degeneration. Am J Ophthalmol. 1995 Sep;120(3):330-41. [PMID: 7661205]
7. Woodward M, Randleman JB, Larson PM. In vivo confocal microscopy of polymorphic amyloid degeneration and posterior crocodile shagreen. Cornea. 2007 Jan;26(1):98-101. [PMID: 17198022]
8. Karp CL, Scott IU, Green WR, Chang TS, Culbertson WW. Central cloudy corneal dystrophy of Francois. A clinicopathologic study. Arch Ophthalmol. 1997 Aug;115(8):1058-62. [PMID: 9258229]
Carroll JN, Maltry AC, Kitzmann AS. Polymorphic Amyloid Degeneration: 73-year-old asymptomatic patient with filamentous corneal opacities. EyeRounds.org. June 29, 2013; available from https://eyerounds.org/cases/173-Polymorphic-amyloid-degeneration.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
