
Decreasing vision
A 41-year-old man returned to the University of Iowa Hospitals and Clinics for an ocular examination. He was followed from infancy through his teenage years and noted to have Axenfeld-Rieger syndrome but had been lost to follow up for many years prior to this presentation. He returned for consultation regarding decreasing vision in his right eye (OD).
Axenfeld-Rieger syndrome without associated glaucoma in both eyes (OU). Born with nystagmus OU and esotropia, prior best-corrected visual acuity (BCVA) 20/80 OD and 20/80 left eye (OS).
Hydrocephalus status post shunt, seizure disorder, mental retardation, dental malformation and jaw malposition, depression.
Phenobarbital, phenytoin, levetiracetam, sertraline
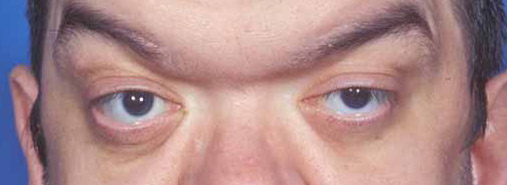 |
The patient was bothered by the decreased vision in the right eye so cataract surgery was planned.
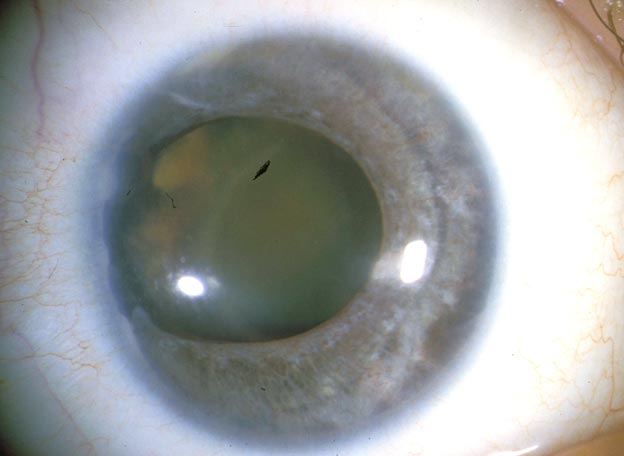 |
The patient underwent cataract surgery OD. Two iris retractors were placed nasally about 60 degrees apart to expand the pupil toward the center of the lens. Trypan blue was used to stain the anterior capsule and facilitate visualization and successful completion of a continuous curvilinear capsulorhexis (CCC). During CCC the zonules in the area of the temporal iris adhesion were felt to be weak. Phacoemulsification using a chopping technique was performed on his dense nucleus without complication. Following careful removal of the residual cortex a 12 mm capsular tension ring was placed to promote capsular stability in the setting of sectoral zonular weakness. An Alcon SA60AT 17.5 diopter lens was placed in the bag and remained centered. A pupilloplasty performed at 2 and 4 o’clock using intraocular scissors shifted the pupil more centrally to keep the visual axis clear of iris tissue.
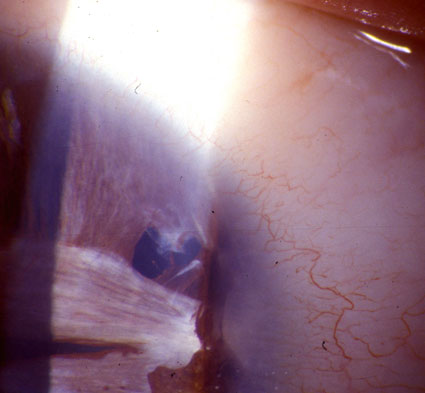
Postoperatively the patient did well and no complications were encountered. Visual acuity improved to 20/100 OD. One year after cataract extraction, the vision was 20/100 OD and 20/300 OS. Intraocular pressure was 19 mmHg OU. The corectopia was stable and visual axis was clear. His posterior chamber intraocular lens remained well centered OD (Figure 3). His left eye had a mature brunescent cataract; however, the patient did not wish to proceed with cataract surgery, as he was satisfied with his current vision.
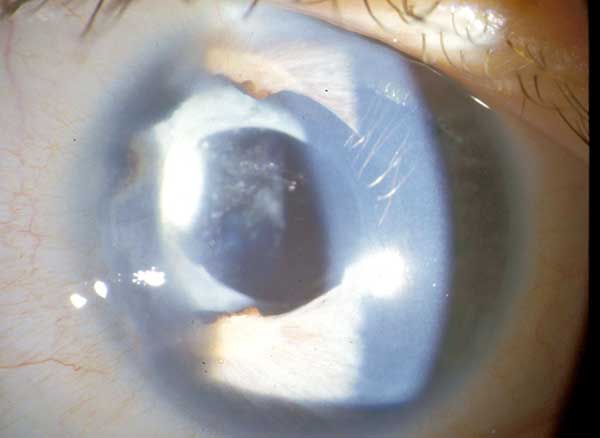 |
Axenfeld-Rieger syndrome (ARS) refers to an autosomal dominant genetic condition characterized by congenital anterior segment dysgenesis and systemic abnormalities. Some literature definitions of ARS attempt to subdivide the disease in the following manner. Axenfeld anomaly: posterior embryotoxon and iris process in the angle but no other iris or systemic findings; Rieger anomaly: embryotoxon, iris process, iris changes without systemic findings; Rieger syndrome: all the above ocular findings plus systemic findings [1,2,4]. Other authors prefer the umbrella terms of anterior segment dysgenesis or iridocorneal dysgenesis.
Clinical findings are bilateral, but often asymmetric. Anterior segment findings can range from subtle to dramatic and include corectopia, polycoria, ectropion uveae, posterior embryotoxon (often with iris strands adherent to the embryotoxon). Gonioscopy should always be performed on these patients, as the embryotoxon may not be visible with slitlamp examination in all cases [3]. Notably, posterior embryotoxon can be a normal finding in up to 10% of the population. Systemic anomalies include craniofacial dysmorophisms such as hypertelorism, telecanthus, maxillary hypoplasia, and a broad, flat nasal bridge. Dental abnormalities include microdontia and hypodontia. Other systemic findings include redundant periumbilical skin, hypospadias, anal stenosis, pituitary abnormalities, and heart defects [4-5]. Given the potential clinical similarity to other causes of anterior segment dysgenesis, careful ocular and systemic evaluation is necessary.
Most cases of Axenfeld Rieger syndrome are inherited in autosomal dominant fashion, but it can also occur sporadically [6]. Development of ARS has been associated with mutations on at least four different gene loci: PITX2 on 4q25, FOXC1 on 6p25, PAX6 on 11p13, and FOXO1A on 13q14 [7-8]. This condition is associated with a 50% risk of developing glaucoma. Most cases are diagnosed in infancy or childhood, but glaucoma typically develops in later childhood or adulthood [9]. Glaucoma is thought to develop as a result of abnormal formation of the trabecular meshwork or Schlemm canal. High iris insertion, rather than the extent of iris defects or iris strands in the angle, seems to correlate more with the development of glaucoma.
Management of these patients should include screening for and management of glaucoma, prevention and treatment of amblyopia, and optimization of lenses for visual function or photophobia and lifelong monitoring of the health of their eyes. These patients should also be monitored by an internist or other specialists as needed, receive good dental care, and have any social and occupational needs addressed.
Like many others with this condition, this patient has had slow progression of his iris defects with an enlarging iris hole OS [3]. Although described in the literature, the progressive changes of the iris in ARS have not been widely demonstrated in photographic format. Though various types of cataract have been documented in ARS, the finding of zonular instability during surgery is not well described. However, this finding is not surprising given that the ocular manifestations of this condition are felt to stem from anterior segment dysgenesis in the third trimester, which could certainly influence lens/zonular development.
| 1979 | 2002 | |
|---|---|---|
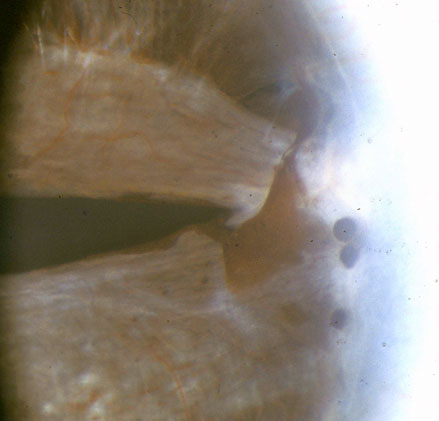 |
 |
 |
Ocular FindingsBilateral, potentially asymmetric Posterior embryotoxon High iris insertion Iris processes in the angle Iris hypoplasia, corectopia, polycoria 50% develop glaucoma |
Systemic FindingsMid-face flattening/maxillary hypoplasia Hypertelorism Telecanthus Broad, flat nasal bridge Hypodontia/microdontia Redundant periumbilical skin Hypospadias |
Differential DiagnosisIridocorneal endothelial syndrome (ICE) – never bilateral! Peter’s anomaly Aniridia Congenital ectropion uvaea Ectropia lentis et pupillae Oculodentodigital dysplasia |
Treatment (for associated glaucoma)Topical medication Systemic medication Surgical intervention Cyclodestructive procedure |
Note: This patient was presented in the literature by Dr. Frank Judisch of the University of Iowa in 1979 prior to the loss to follow up.[10]
Rogers GM, Longmuir SQ, Oetting TA. Axenfeld-Rieger Syndrome and Cataract. EyeRounds.org. July 29, 2013; available from https://eyerounds.org/cases/174-Axenfeld-Rieger.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
