
Chief Complaint: Decreased vision in both eyes
A 17-year-old male was referred to the University of Iowa Hospitals and Clinics (UIHC) Neuro-Ophthalmology Service by his local optometrist for evaluation of severe, painless, bilateral vision loss. Four months prior to presentation, the patient had normal uncorrected vision and passed his driver’s license vision screening. He first noted vision difficulties when trying to follow the flight of a golf ball two months prior to presentation and later had trouble with reading. The patient could not recall a specific moment of worsening and he was unsure whether one eye was affected prior to the other or if there was synchronous involvement.
Normal in all fields of gaze
Moderate acne, large fibroma-like lesions on nose
 |
 |
| right eye | left eye |
 |
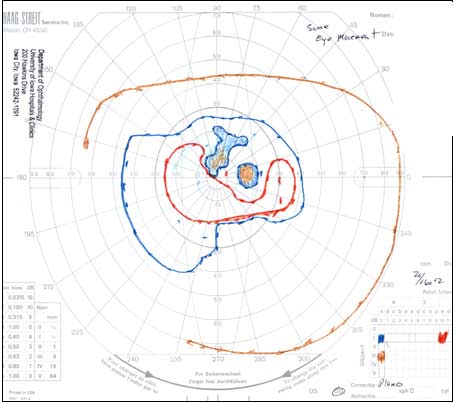 |
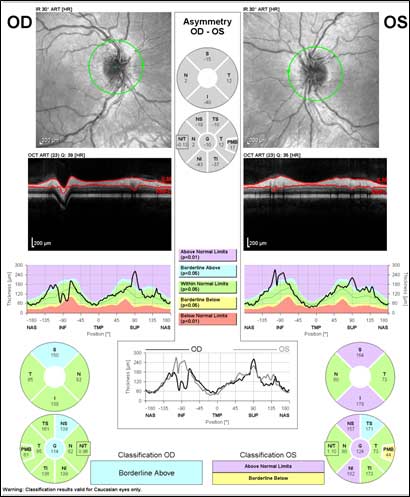
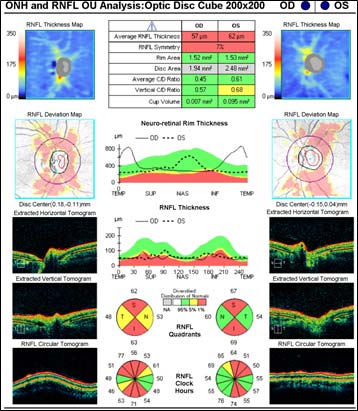
Figure 3: Optical coherence tomography (OCT) of optic nerve
Magnetic resonance imaging (MRI) of the brain/orbits with and without contrast:
The patient had a bilateral optic neuropathy with central scotomas in each eye. Despite an unclear history of sequential vision loss between the two eyes and a lack of family history for visual loss, the appearance of the optic nerves, painless bilateral involvement, young age, and male gender was suspicious for Leber hereditary optic neuropathy (LHON). The mild optic nerve enhancement on the MRI could be compatible with an inflammatory optic neuropathy, but given the overall clinical picture, LHON was thought to be more likely and therefore genetic testing was elected.
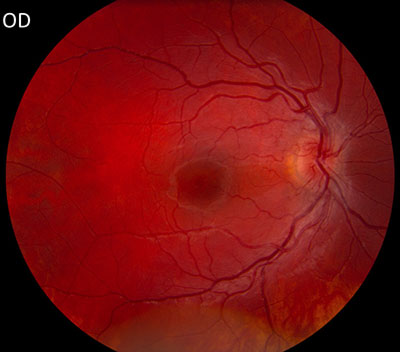
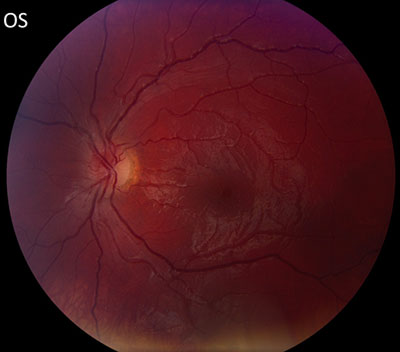
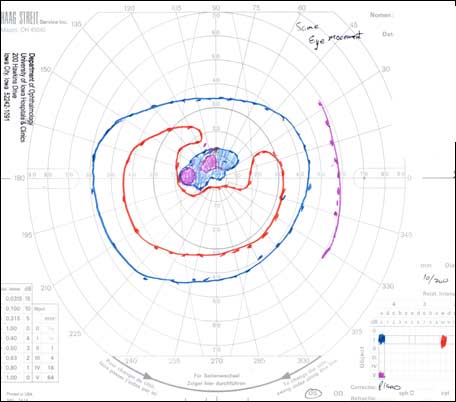
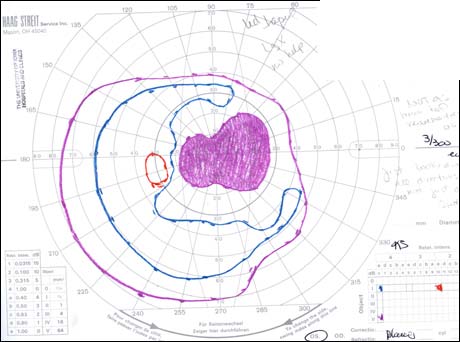
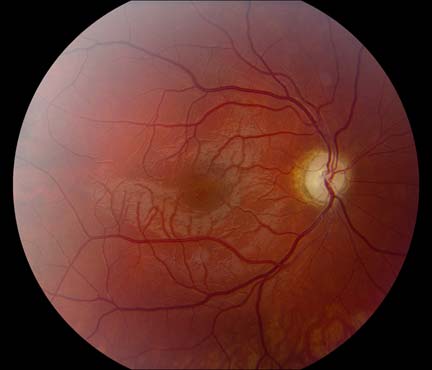
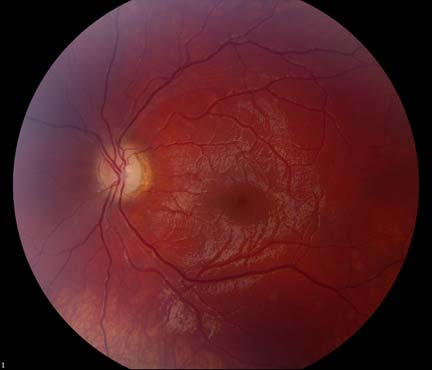
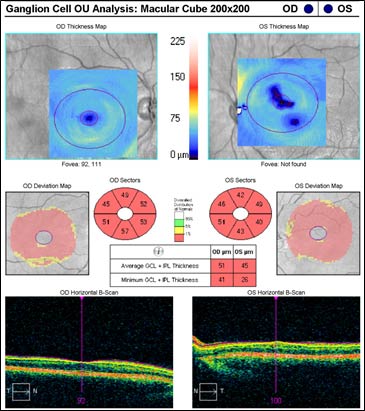
The patient underwent genetic counseling and testing for LHON at the University of Iowa Molecular Ophthalmology Laboratory. His genetic testing came back positive for the 11778 mitochondrial DNA point mutation confirming the diagnosis of LHON. During continued follow-up over the next year, the patient’s visual acuity continued to worsen, then stabilized with count fingers (CF) at one foot bilaterally. In addition, his visual fields worsened with progression to dense, large central scotomas OU (Figure 4). Fundoscopic exam revealed the development of temporal optic nerve pallor with loss of hyperemia OU (Figure 5). OCT showed thinning of the RNFL in the maculopapillary bundle, continued thickening in the arcuate bundles, and diffuse thinning of the ganglion cell layer, consistent with LHON (Figure 6). Over the course of several years, his central scotomas decreased mildly; however, he remained CF at one foot.
 |
 |
 |
 |
 |
 |
LHON was the first human disease in which mitochondrial (mt) DNA point mutations were identified as the causative etiology (Wallace et al., 1988). Over 95% of cases can be attributed to three mtDNA pathogenic point mutations, 11778/ND4, 3460/ND1, and 14484/ND6 (Riordan-Eva et al., 1995). These mutations affect complex I subunit genes in the mitochondrial electron transport chain (Carelli et al., 2004). Since its discovery, LHON has been recognized as one of the most common mitochondrial diseases with a prevalence of 1 in 30,000 to 1 in 50,000 people depending on the population (Puomila et al., 2007). The disease is maternally inherited and demonstrates incomplete penetrance, primarily affecting males in their second and third decades of life.
The classic ophthalmologic manifestation of LHON is an acute/subacute painless loss of central vision, which is typically sequential, manifesting in one eye and then proceeding to affect both eyes symmetrically within weeks to months (Yen et al., 2006; Smith et al., 1973). The vision loss is usually profound with visual acuities of 20/200 or worse. Degeneration of visual acuity occurs over a period of days to weeks and the median inter-eye delay is eight weeks (Harding et al., 1995). The visual acuity loss stabilizes within one year and recurrence of the disease is rare. Although sequential presentation is much more common, the disease process can present with nearly synchronous involvement of both eyes. True unilateral ocular involvement is rare (Nagai et al., 2005). The central vision deficit corresponds with the selective degeneration of retinal ganglion cells (RGCs) and the preferential loss of small-caliber fibers in the papillomacular bundle (Sadun et al., 2000). Other ophthalmologic manifestations of LHON include dyschromatopsia, diminished contrast sensitivity, and a bilateral dense central/centrocecal scotoma on visual field testing (Newman et al., 1991; Fraser et al., 2010).
Although most affected patients remain legally blind, some patients demonstrate a milder disease or demonstrate some degree of spontaneous recovery with decrease in the size of the central scotoma or development of small islands of vision within the scotoma (fenestrations) (Mackey et al., 1991).
Major visual prognostic factors include type of pathogenic mutation, age of onset, and optic disc size. The most common mutation, 11778/ND4, is associated with the worst visual prognosis and lowest visual recovery rate (Stone et al., 1992). A rarer mutation, 14484/ND6, results in a milder course and higher incidence of spontaneous partial visual recovery. Patients carrying the 14484/ND6 mutation have demonstrated late recovery up to five years after disease onset (Johns et al., 1993). Younger age of onset has also been associated with a milder disease course and increased spontaneous visual recovery. It has been noted that the visual prognosis of childhood LHON is better than adults for each of the mutations (Barboni et al., 2006). A large optic disc has been correlated with increased spontaneous visual recovery and an overall better visual outcome compared to patients with a smaller optic disc. This is thought to be due to the crowding of the axons of retinal ganglion cells in a small optic disc that makes the nerve more susceptible to optic neuropathy, similar to the well documented “disc at risk” in anterior ischemic optic neuropathy (Ramos et al., 2009).
Interestingly, many individuals carrying a pathologic mutation are asymptomatic. It is believed that the presence of a pathogenic mtDNA LHON-causing mutation is necessary but not sufficient for the development of LHON. Of those harboring the pathogenic mutations, approximately 50% of males and 10% of females undergo clinical manifestations of visual loss (Yen et al., 2006; Fraser et al., 2010). Triggering factors have been proposed to contribute to clinical conversion from the asymptomatic to the symptomatic stage, including environmental factors (alcohol, tobacco, viral infections, toxin exposure, mitotoxic medications), genetic factors (nuclear modifier genes, mtDNA haplogroups), and hormonal factors (testosterone, low estrogen) (Sadun et al., 2003; Hudson et al., 2007).
LHON has traditionally been characterized into three major phases: asymptomaticacute/subacute, and atrophic. During the asymptomatic phase, patients enjoy relatively good visual acuity; however, ophthalmoscopic examination may reveal circumpapillary vessel tortuosity and capillary microangiopathy in some asymptomatic patients (Nikoskelainen et al., 1983). Furthermore, OCT of the optic nerve may demonstrate an increased RNFL thickness of the temporal quadrant in some patients, which corresponds with the location of the papillomacular bundle (Savini et al., 2005a). This increased thickness is thought to represent axoplasmic flow stasis and edema as a result of RGC injury, and suggests that there is selective and early involvement of the papillomacular bundle in LHON even in patients that are asymptomatic. It is currently unknown whether this RNFL swelling heralds future clinical conversion.
The acute/subacute phase results in profound vision loss and a classic triad on fundoscopic examination characterized by circumpapillary telangiectatic microangiopathy, swelling around the disc (pseudoedema), and lack of leakage on fluorescein angiography (Smith et al., 1973; Nikoskelainen et al., 1977). Longitudinal OCT studies of the optic nerve typically demonstrate significantly increased RNFL thickness in all of the quadrants except the temporal quadrant during the acute/subacute phase (Savini et al., 2005b). The less significant increase in the temporal RNFL is thought to be due to the preceding atrophy of the papillomacular bundle that occurs in the asymptomatic phase. Fundoscopic examination at three months demonstrates temporal pallor and disappearance of the triad observed in the acute/subacute phase (Carelli et al., 2004).
At six months after onset of the acute/subacute phase, the patient enters the atrophic phase. Fundoscopic examination reveals diffuse pallor of the disc with no signs of edema. OCT shows a significant reduction in RNFL thickness in all quadrants. Within one year, most patients experience stabilization of their vision, but are often left with severe bilateral vision deficits and are legally blind. Recovery of vision is unlikely at this point in the disease process.
Although a majority of LHON patients demonstrate normal MRI findings, various imaging abnormalities have been described over the disease course. During the acute phase, optic nerve enlargement and enhancement have been documented, complicating the diagnosis of LHON versus optic neuritis, as demonstrated in our case (Kermode et al., 1989; Mashima et al., 1998; Vaphiades et al., 2003; Phillips et al., 2003). Multiple sclerosis-like white matter lesions have also been observed (Harding et al., 1992). The atrophic phase may demonstrate retrochiasmatic tissue loss with reduction of grey matter volume in the primary visual cortex and reduced white matter volume in the optic chiasm, optic tract, and optic radiations (Barcella et al., 2010).
Genetic testing for the three most common disease point mutations is required for definitive diagnosis in the context of clinical manifestations concerning for LHON. Genetic testing for mutations in LHON is available through the Carver Laboratory for Molecular Diagnosis (www.carverlab.org), a nonprofit laboratory dedicated to providing molecular diagnostic studies for heritable eye conditions. Other sources for genetic testing of LHON can be found at www.genetests.org. However, 10% of mitochondrial mutations responsible for LHON will be missed by screening of the three most common disease point mutations, so some cases of LHON remain a clinical diagnosis.
It is widely accepted that dysfunction of complex I in the energy transport chain results in increased production of reactive oxygen species. The increased oxidative stress could ultimately result in apoptosis of susceptible RGCs in LHON, especially the parvocellular RGCs that give rise to the papillomacular bundle. Therefore, treatment of LHON has primarily focused on exploring antioxidants as possible therapeutic agents. The therapeutic benefit of a number of antioxidants has been explored in the past including cyanocobalamin, folic acid, ascorbic acid, and coenzyme Q10, all of which have been disappointing without demonstrable benefit (Rosenfeld and Sadun, 2010). However, recent studies have shown that two antioxidants, idebenone and α-tocotrienol-quinone (EPI-743), may improve the final visual outcome in affected patients. Both compounds are believed to work by carrying electrons directly to complex III and thus bypassing the deficient complex I.
Interest in idebenone, a quinolone, as a potential therapeutic agent in the treatment of LHON has been present for the past decade. Anecdotal reports in the past have suggested variable results. Recently, the results of the first controlled clinical trial and a large retrospective analysis were published (Klopstock et al., 2011; Carelli et al., 2011), which both showed partial effectiveness in ameliorating the final visual deficits in LHON-patients. This was primarily accomplished by increasing the rate of visual recovery after the acute phase. Therefore, it is currently accepted that in LHON-patients with relatively recent disease onset, treatment with high dose (300 mg three times daily) idebenone may benefit some patients. The compound has been documented to have a relatively safe side-effect profile. There is no current recommended duration of therapy; however, early initiation of treatment seems to have the best chance of improving the visual outcome. Furthermore, it may be prudent to avoid drugs implicated with mitochondrial dysfunction in patients with a family history suspicious for LHON or other mitochondrial diseases (Nadanaciva and Will, 2011).
A second drug, the antioxidant α-tocotrienol-quinone (EPI-743), a vitamin E derivative, has also shown promising early results. A small open-label study of five patients with acute LHON showed that early treatment with EPI-743 arrested disease progression in four out of five the LHON-patients (Sadun et al., 2012). However, a double-blind, randomized placebo-controlled trial is needed to provide confirmation of EPI-743 in the treatment of acute LHON.
Gene therapy for LHON is currently being studied. Development of gene therapies have been complicated by difficulties in introducing genes coding for functional complex I subunits into the mitochondria of affected RGCs. Genetic heterogeneity has also impeded the advancement of this approach. However, several recent studies in animal models have resulted in successful introduction of functional complex I subunit proteins into RGC mitochondria (Guy et al, 2009; Koilkonda et al., 2009; Shi et al., 2012).
Epidemiology
|
Signs
|
Symptoms
|
Treatment
|
Baldwin A, Chen JJ, Kardon RH. Leber Hereditary Optic Neuropathy: A 17-year-old male presents with progressive, painless, bilateral vision loss. EyeRounds.org. June 25, 2013; available from https://eyerounds.org/cases/172-LHON.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
