
Corneal epithelial-stromal and stromal dystrophies are a group of inherited disorders of the cornea that are caused by progressive accumulation of deposits within the layers of the cornea. These deposits are not caused by inflammation, infection, or trauma, but by genetic mutations that lead to transcription of aberrant proteins resulting in the accumulation of insoluble material within the cornea. The disorders may or may not affect vision and may or may not be symmetrical (1). The 2015 International Committee for Classification of Corneal Dystrophies (IC3D) classification system has divided corneal dystrophies into 4 categories: epithelial and subepithelial dystrophies, epithelial-stromal dystrophies, stromal dystrophies, and endothelial dystrophies. Most dystrophies previously considered stromal are now classified as either epithelial-stromal dystrophies or stromal dystrophies. Table 1 and 2 list the epithelial-stromal dystrophies and stromal dystrophies (2). The old classification for corneal stromal dystrophies is listed in Table 3.
| Table 1: Epithelial-stromal Corneal Dystrophies |
|---|
|
| Table 2: Stromal Corneal Dystrophies |
|---|
|
Table 3. Old classification of corneal stromal dystrophies |
|---|
|
Epithelial-stromal dystrophies are caused by mutations in transforming growth factor beta-induced (TGFβI) gene, also known as the BIGH3 gene. TGFβI is located on chromosome 5q31 and codes for keratoepithelin, a protein secreted by corneal epithelium. This protein acts as an adhesion protein and is present in normal stroma. Being a small protein roughly the size of albumin, it has the capability to diffuse through the corneal stroma. When a mutation in the TGFβI gene occurs, the keratoepithelin structure is abnormal and accumulation of the insoluble protein or its proteolytic fragments occurs in the cornea (1, 3). Interestingly, the TGFβI gene mutation was discovered in part at the University of Iowa. A group of researchers and clinicians including Edwin M. Stone, Robert Folberg, and Jay H. Krachmer mapped granular type I, granular type II, and lattice dystrophy to chromosome 5q in 1994 (4). To date, 63 different mutations have been identified in the TGFβI gene. No effective treatments to prevent or attenuate the deposition of the keratoepithelin have been identified. The dystrophies typically have an autosomal dominant inheritance and involve Bowman layer and stroma (3).
Reis-Bücklers, formerly known as Granular corneal dystrophy type III or Corneal Dystrophy of Bowman's type I, typically present with normal corneas at birth but develop painful recurrent erosions, opacification, and progressive vision loss within the first decade of life (1). Irregular, grey-white, geographic-like opacities are located in the Bowman layer and anterior stroma. In more advanced stages of the disease, the opacities can extend to the limbus and deeper stroma (2). Histopathology reveals anterior stromal and subepithelial deposits of hyaline-like material which disrupt and often replace Bowman's layer (See Figure 1A and 1B). The deposits stain red with Masson trichrome stain (2). The hyaline-like material consists of rod-like bodies ultrastructurally, which helps distinguish it from Thiel-Behnke corneal dystrophy (1, 2).
| A: H&E of Reis Bückler showing destruction of Bowman's layer and irregular epithelium | B. Masson Trichrome stain demonstrating epithelial staining |
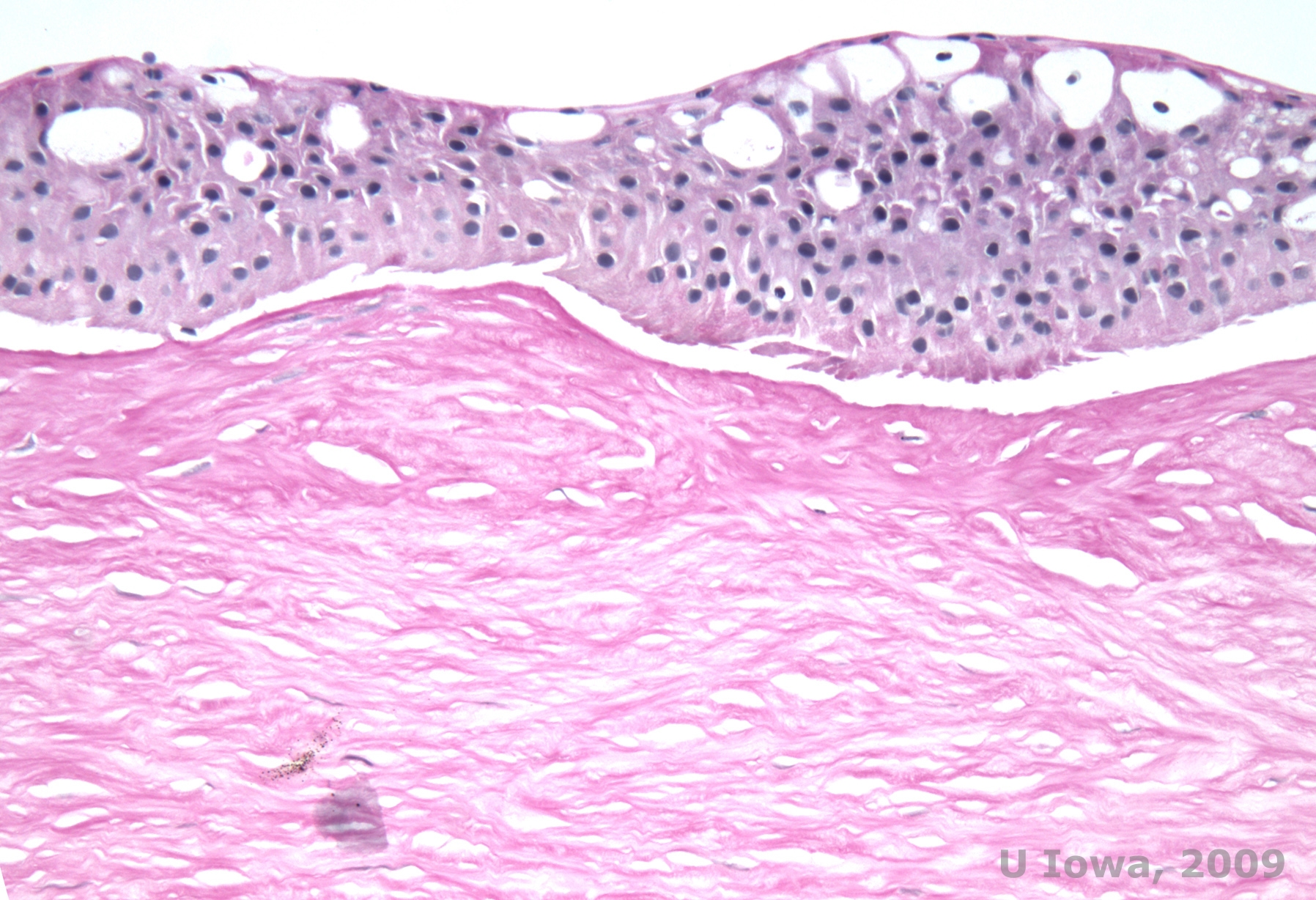 |
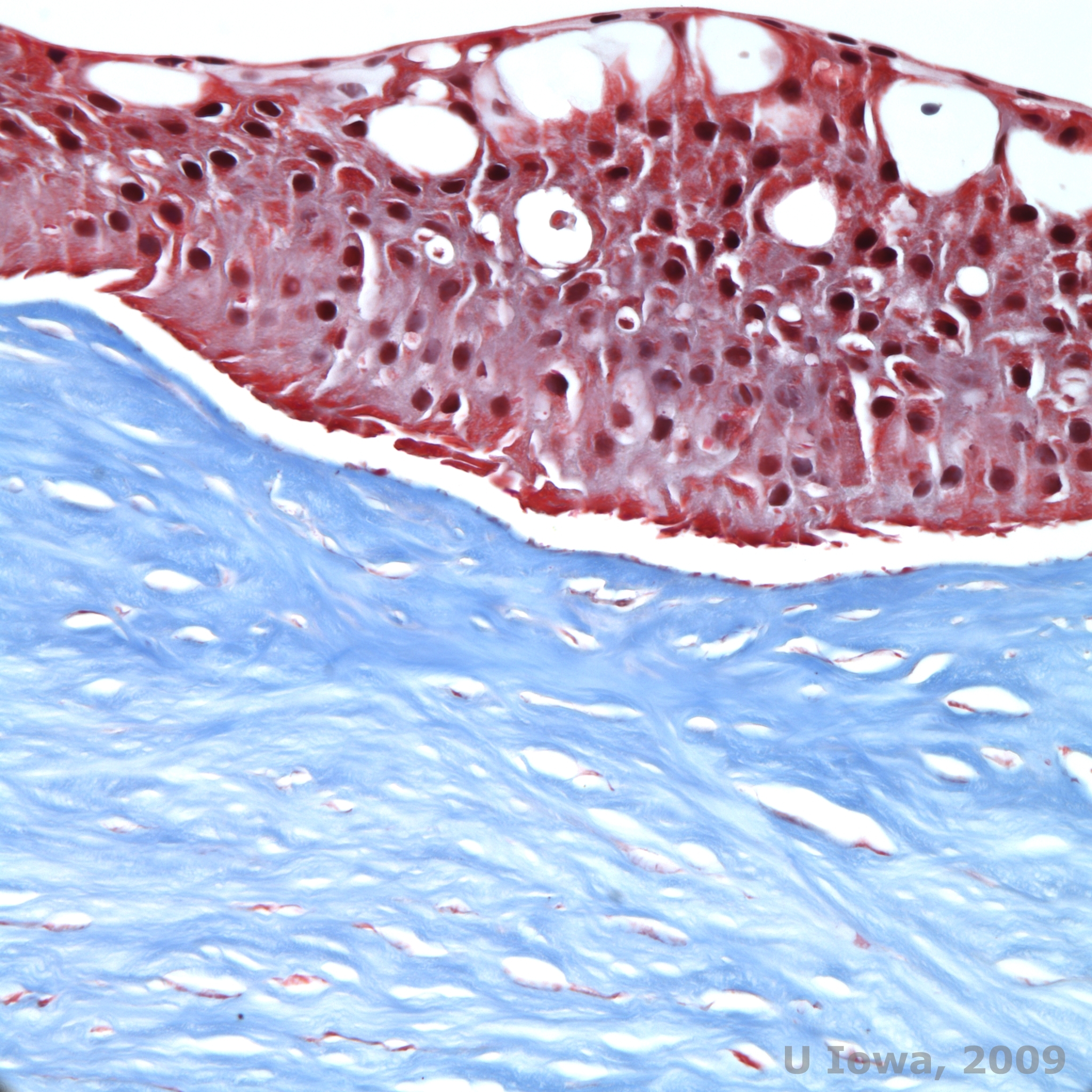 |
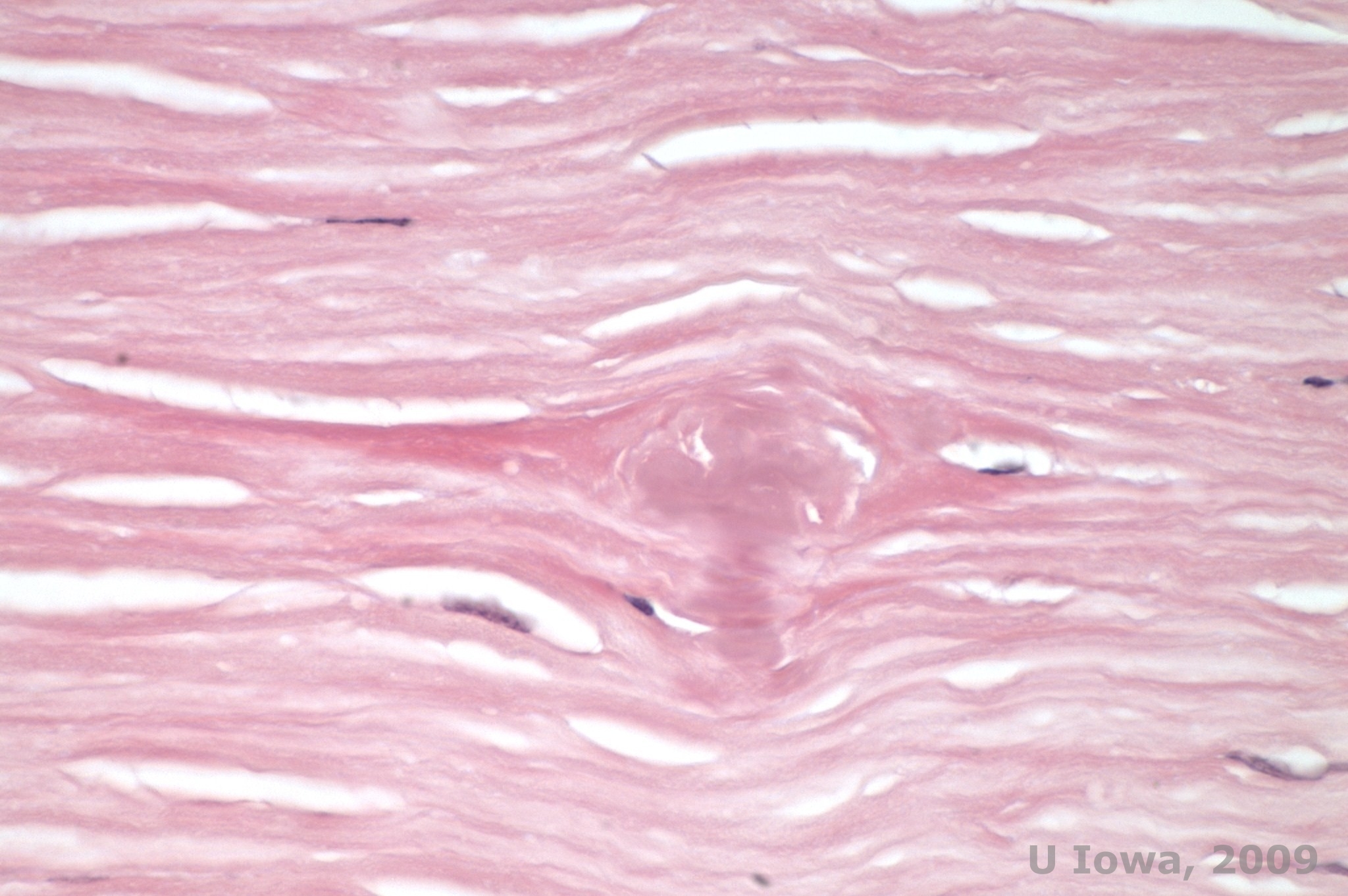
Lattice corneal dystrophy (LCD) is the most common of the corneal epithelial-stromal dystrophies. It is typically an autosomal dominant, bilateral disease that typically presents toward the end of the first decade of life with symptoms of recurrent corneal erosions and decreased vision. It is characterized by lattice lines which are linear, radially oriented, branching refractile opacities described as "glass like" located in the anterior stroma (See Figure 2A and 2B). These lattice lines are initially found in the superficial central cornea. As the disease progresses, they spread deeper and peripherally in the stroma with sparing of the limbus (1, 2). Other exam findings include fleck-like opacities, subepithelial white dots, and "ground-glass" stromal haze, which starts centrally and become more diffuse (2). Many patients with LCD will require surgical intervention for treating recurrent erosions and decreased vision. If the disease is located anteriorly in the stroma, patients can often be successfully treated with phototherapeutic keratectomy (PTK). Some require corneal transplantation. Because keratoepithelin, the protein produced by the TGFβI gene, is produced mostly in the corneal epithelium, the disease tends to recur in corneal grafts (1).
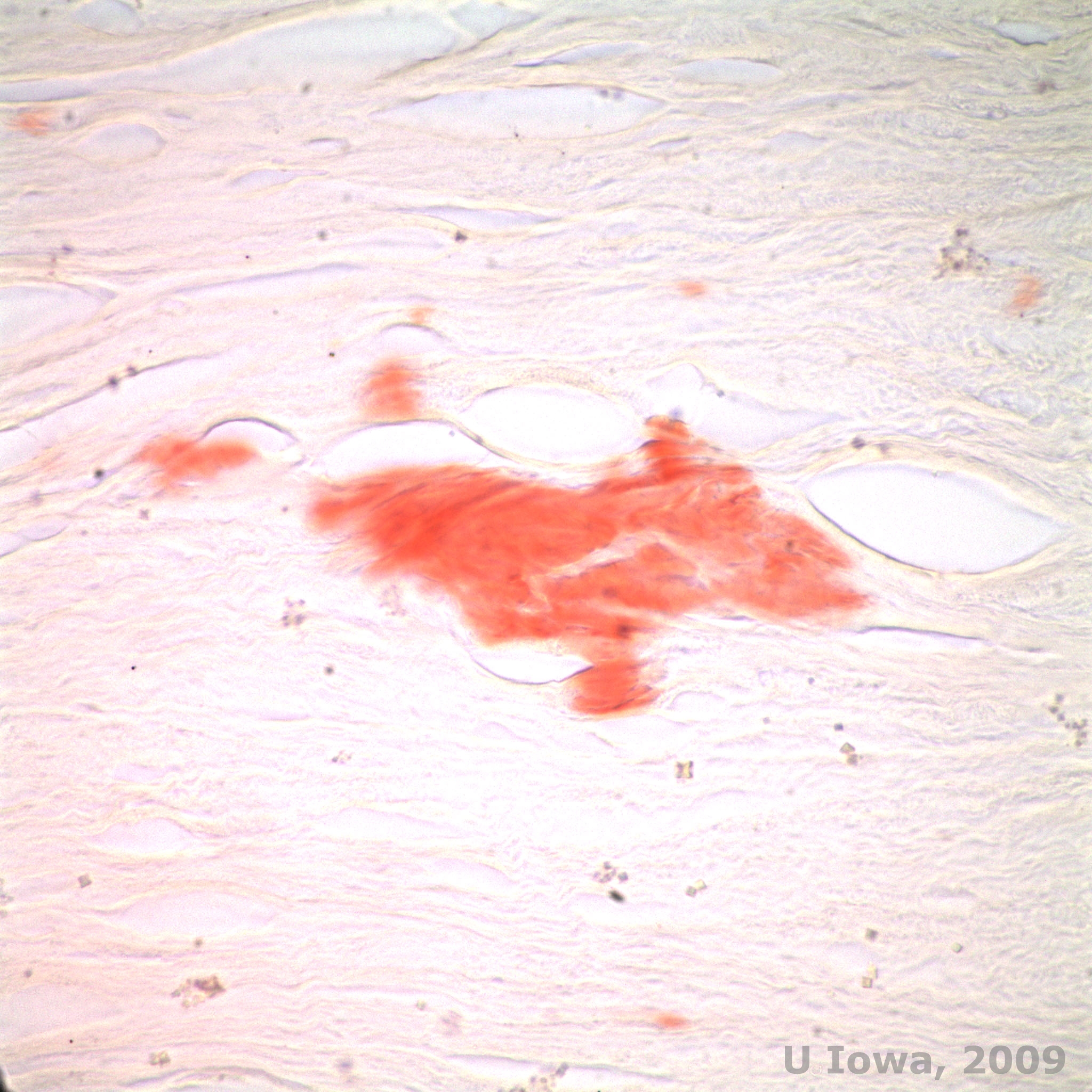
In LCD, amyloid deposits accumulate between the epithelial basement membrane and Bowman layer as well as in the stroma, causing distortion of the lamellar architecture. The deposits stain positively with immunohistochemistry using antibodies against keratoepithelin (2). The deposits appear as amorphous pink deposits on hematoxylin and eosin (H&E) stain (See Figure 1C and 1D) and stain with Congo red stain demonstrating the classic apple green birefringence on cross-polarization (See Figure 2E and 2F) (1). Absence or thinning of Bowman layer, epithelial atrophy and basal epithelial degeneration can also be found on histopathology in LCD (2).
LCD type I is the classic form of LCD caused by a mutation in the TGFβI gene resulting in isolated amyloid deposition in the cornea. Four LCD variants had been identified: LCD type IIIA, type I/IIIA, type IV, and polymorphic amyloidosis. LCD variants present later in life than classic LCD. LCD type IIIA presents in the 5th-7th decade, usually with epithelial erosions. It has thicker lattice lines, described as "ropy-appearing", which extends to the limbus. LCD type I/IIIA has thin lattice lines. LCD type IV presents in the 7th-9th decade with small lattice lines. Amyloid deposits in LCD type IV are found in the deep stroma and epithelial erosions rarely occur. Lattice lines are absent in polymorphic amyloidosis type [http://webeye.ophth.uiowa.edu/eyeforum/cases/173-Polymorphic-amyloid-degeneration.htm] and rarely do epithelial erosions occur (2).
LCD type II is a systemic amyloidosis syndrome known as Meretoja syndrome [http://webeye.ophth.uiowa.edu/eyeforum/cases/176-meretoja.htm]affecting the skin, cranial nerves and cornea. It presents in early adulthood with peripheral neuropathies, cranial neuropathies, hound-like facies, dry skin, blepharochalasis, protruding lips, and corneal lattice lines. This type has been linked to the gelsolin gene on chromosome 9, which encodes for an amyloid precursor protein which functions to remove actin from sites of injury and inflammation (1). The name is a misnomer and is not considered a variant of lattice corneal dystrophy (2).
| A: Left eye on retroillumination demonstrating anterior stromal deposits in lattice corneal dystrophy | B: Left eye with higher power showing linear anterior stromal deposits. |
 |
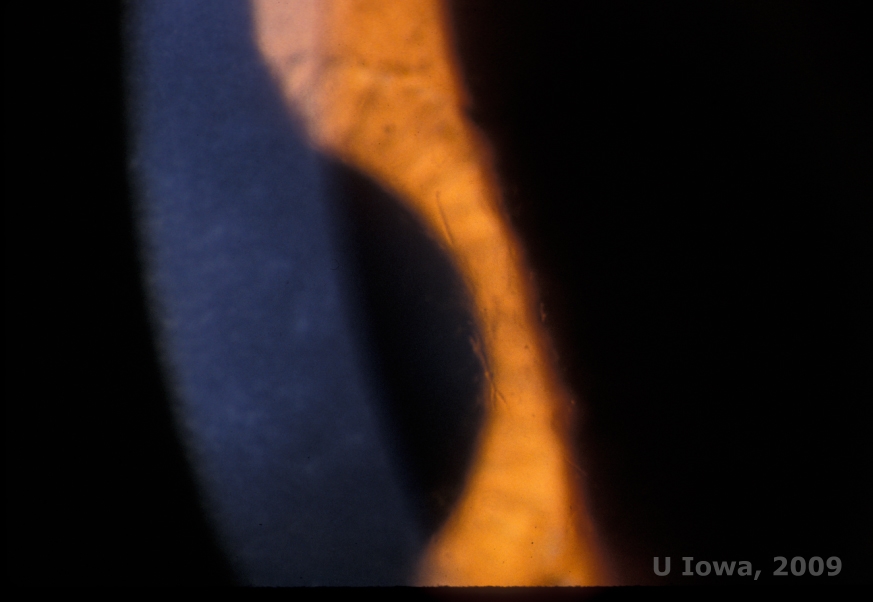 |
| C: H&E stain of cornea with lattice. Note pink amorphous deposits in stroma | D: A closer view of the pink, amorphous deposits |
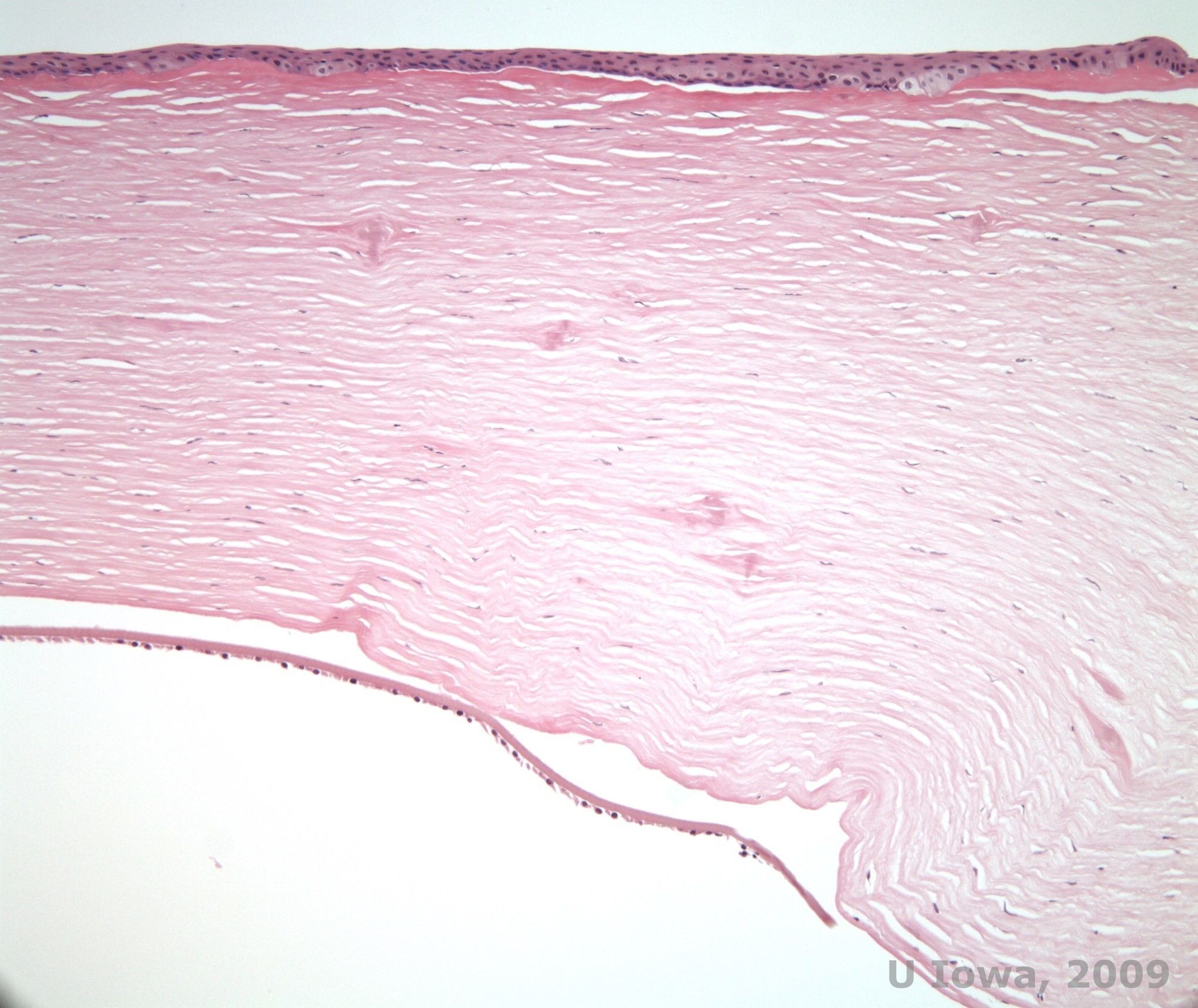 |
 |
| E: Congo red stain, highlighting amyloid | F: Apple-green birefringence of amyloid with cross-polarization. |
 |
 |
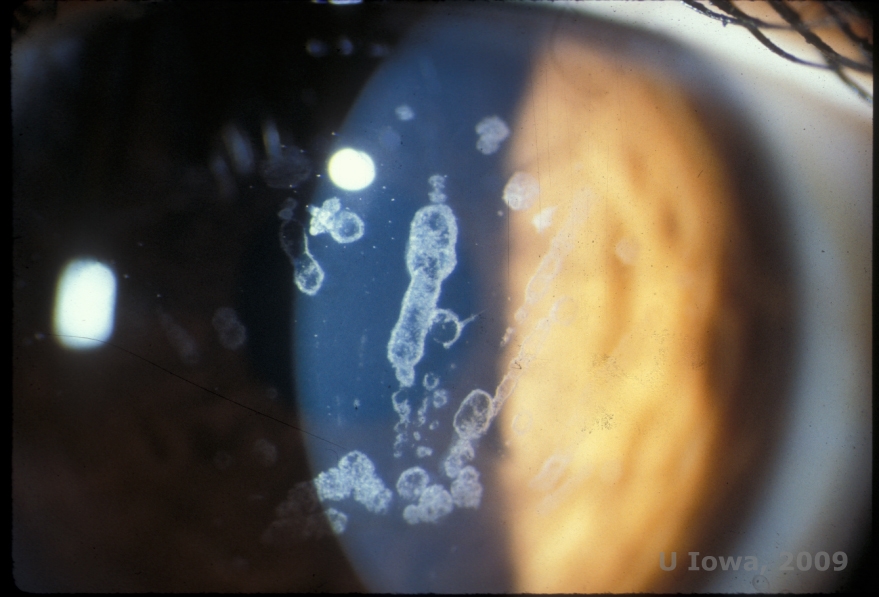
Granular corneal dystrophy, type I (GCD1) is a bilateral, autosomal dominant disease associated with a mutation in the TGFβI gene that leads to the deposition of a hyaline material in the corneal stroma. It typically presents early in the first decade of life with gray-white, "crumb-like" opacities in the anterior to mid stroma, extending into the posterior stroma in advance disease (1, 2). These opacities are discrete deposits located centrally, with clear cornea located in the periphery and clear cornea between deposits (See Figure 3A and 3B). The disease is typically asymptomatic early on, but with time the opacities can coalesce and lead to decreased vision. Recurrent corneal erosions can occur in GCD but at a lower incidence than in LCD (1, 5). Patients can also experience glare and photophobia (2). Treatment early on in the disease process is often observation only. However, as the disease progresses, PTK and corneal transplantation may be needed to improve vision and erosion symptoms. Like LCD, the disease can recur in corneal grafts.
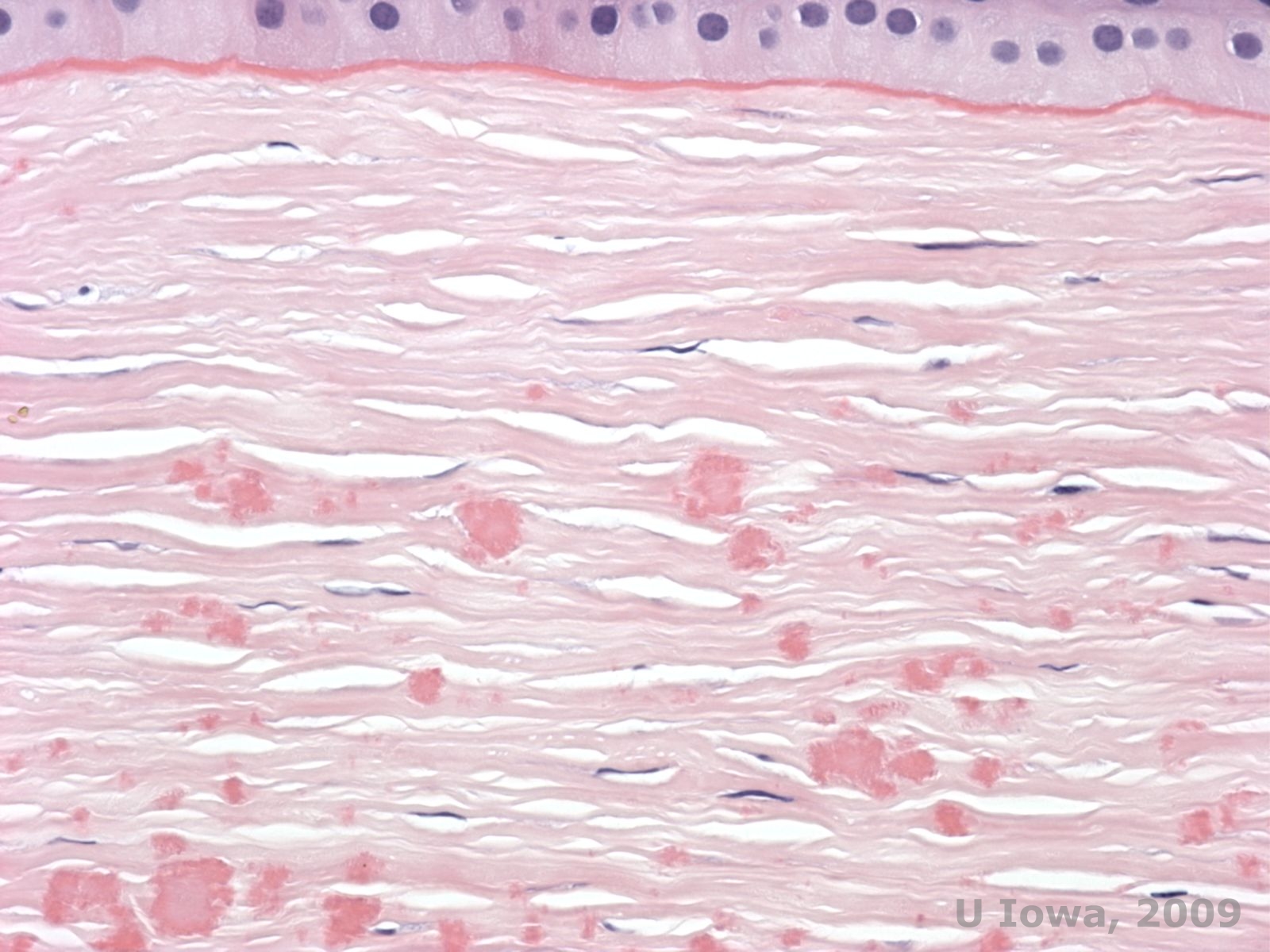
Histopathologically the opacities are eosinophilic deposits often described as "rock candy like" in the anterior stroma made of a hyaline-like material. With time, the deposits progress into the deeper corneal stroma. The hyaline material stains bright red with Masson trichrome stain (See Figure 3C and 3D).
| A:Slit lamp photo of Granular Corneal Dystrophy, Type I | B: Note the "crumb-like" stromal deposits with clear intervening stroma. |
 |
 |
| C: H& E stain of cornea showing eosinophilic "rock candy like" hyaline deposits in stroma | D: Hyaline material stains bright red with Masson-Trichrome |
 |
 |
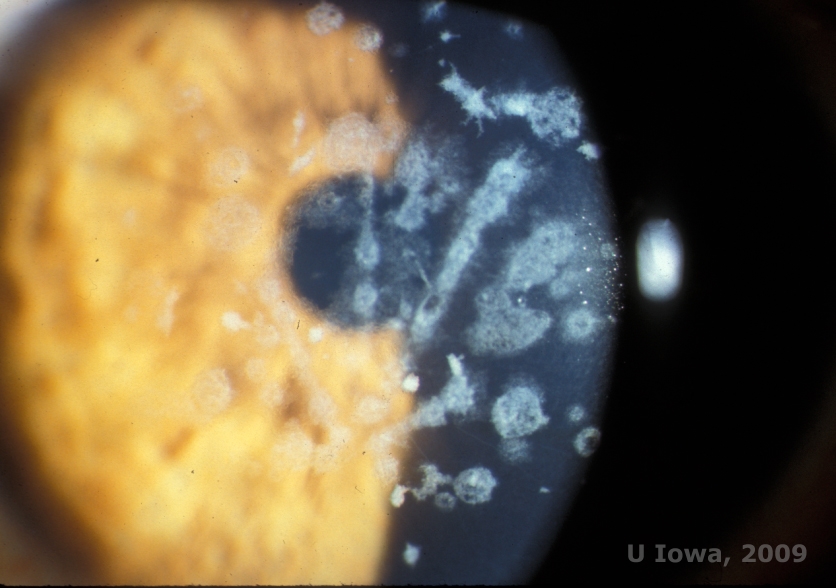
Granular corneal dystrophy, type II (GCD2), formerly known as Avellino or combined granular-lattice corneal dystrophy, is an autosomal dominant disease linked to a mutation in the TGFβI gene that leads to a deposition of both hyaline and amyloid in the corneal stroma. Typically, patients present in the second decade of life with small grey-white dots in the superficial stroma. The opacities can also appear thorn-like, ring, or stellate in shape. In retroillumination, they are partially translucent. Later in the disease process, they can develop lattice lines as well (See Figure 4A and 4B). These lines do not cross each other and appear whiter and less refractile than lattice lines. Symptoms of GCD2 are pain with epithelial erosions and visual impairment (2).
Histopathologically, the cornea will have stromal deposits that stain red with Masson Trichrome, indicating the presence of hyaline (See Figure 4C). In addition, staining with Congo red will demonstrate apple-green birefringence on cross-polarization indicating the presence of amyloid (See Figure 4D). The disease was thought to have originated from a family in Avellino, Italy. However, GCD type II has now been reported in patients from many other countries as well (2,5), with the highest prevalence being in east Asia.
| A: Avellino dystrophy showing lattice like and granular like deposits in corneal stroma | B. Masson Trichrome stain demonstrating anterior stromal hyaline deposits |
 |
 |
| C: Congo red stain showing pink, amorphous amyloid deposits in the same corneal specimen. | D: Cross-polarization reveals apple green birefringence indicating amyloid. |
 |
 |
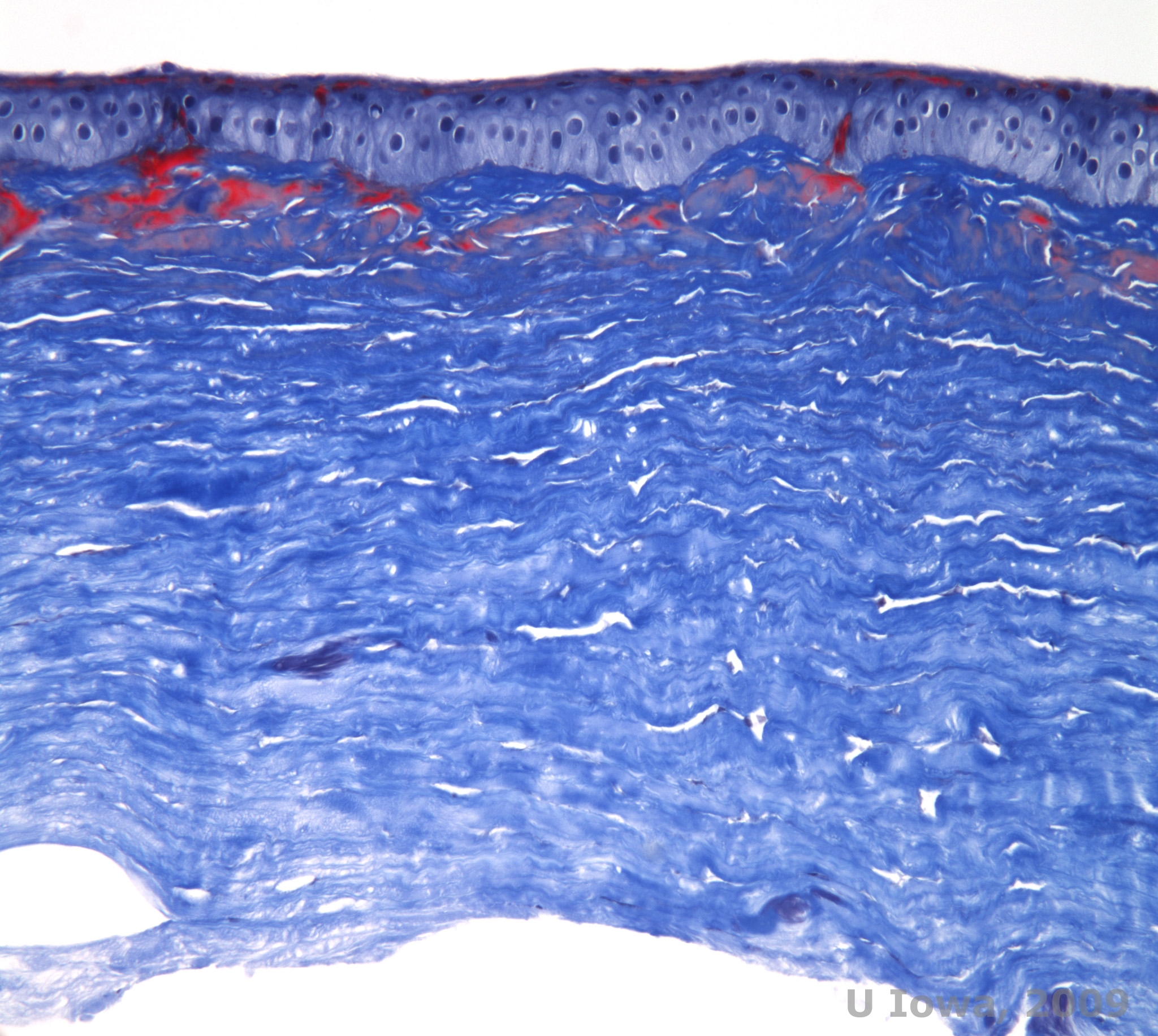
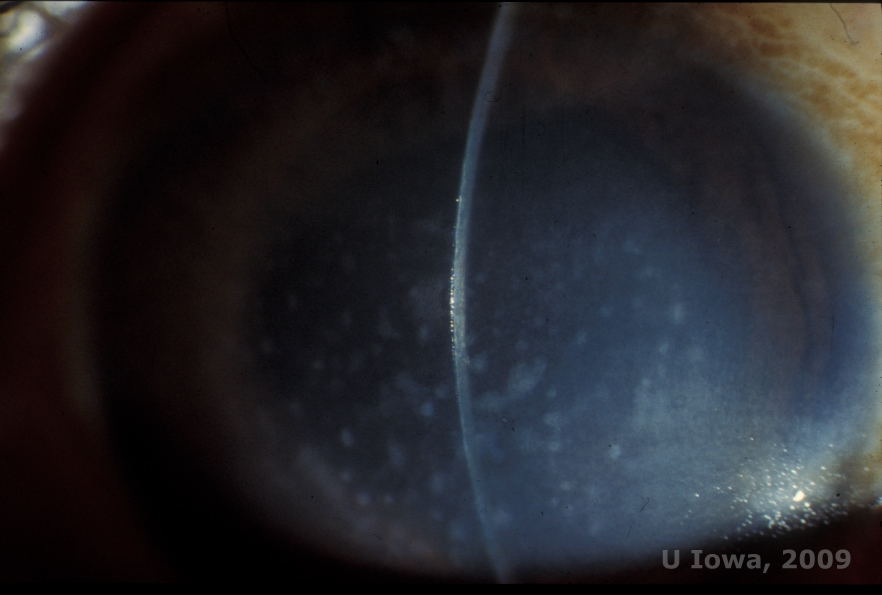
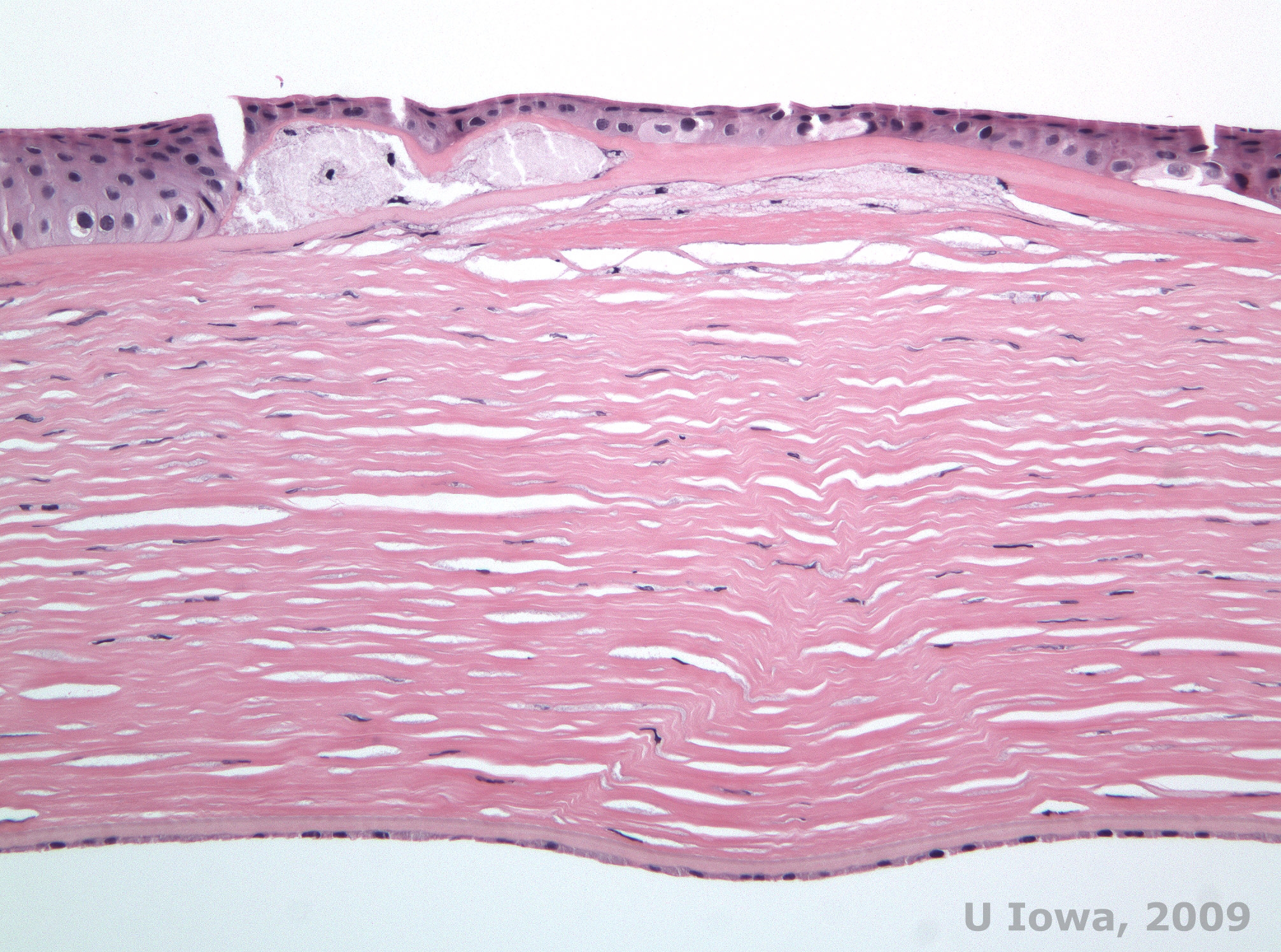
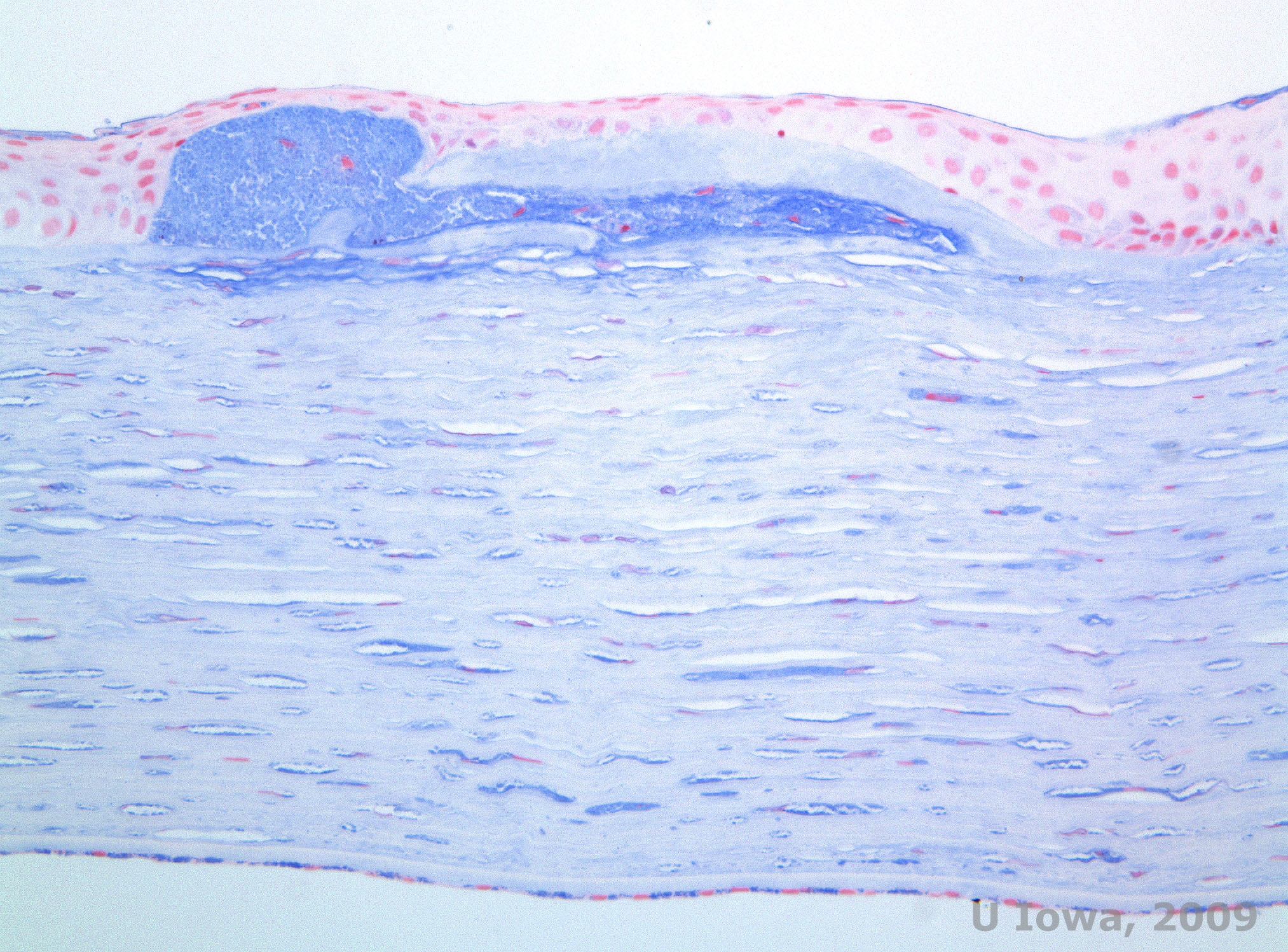
Macular corneal dystrophy (MCD) is an autosomal recessive disease caused by a mutation in carbohydrate sulfotransferase 6 gene (CHST6) on chromosome 16 that leads to a defect in the synthesis of keratan sulfate, the major glycosaminoglycan of the cornea. It is the less common than LCD or GCD, but tends to impact vision more severely. Although MCD is less common worldwide than LCD or GCD, it is the most common of the corneal stromal dystrophies in places such as Iceland and Saudi Arabia (2,6). Gray-white, fleck-like anterior stromal lesions similar to GCD1 appear in the cornea in the first decade of life. Unlike GCD1, however, there is stromal haze between the deposits and the entire cornea from limbus to limbus is often involved (See Figure 5A and 5B). The cornea is thin and as the disorder progresses Descemet membrane becomes grey and develops guttae. Epithelial erosions can occur, but less in MCD than in LCD. Patients typically develop severe visual loss by the second to third decade of life due to diffuse corneal haze. PTK can be performed in some early cases of MCD. However, this condition is generally not as amenable to PTK as lattice or granular dystrophy and often requires corneal transplantation for treatment (7). Recurrence in grafts is less common in MCD than with granular or lattice dystrophy (1,2,5,6,8).
The stromal deposits in MCD are composed of mucopolysaccharides that accumulate within the endoplasmic reticulum of keratocytes of the corneal stroma, extracellularly between stromal lamellae, and within the epithelium, Descemet membrane and endothelium. These deposits stain blue with Alcian blue (See figure 5C and 5D) (1). There are breaks in Bowman layer and guttae with thickening of Descemet membrane (2).
Three subtypes of MCD have been described based on the presence or absence of immunoreactive keratan sulfate within various tissues. Type I does not have immunoreactive keratan sulfate in the corneal stroma, keratocytes, sera or cartilage, and is the most common variant of MCD worldwide. Type IA lacks keratan sulfate in the stroma, sera, and cartilage, but has detectable levels inside keratocytes. Type II has keratan sulfate present at much reduced levels in the stroma, keratocytes, sera and cartilage (6).
| A: Slit lamp photo of macular corneal dystrophy. | B: Note the haze between the corneal stromal deposits |
 |
 |
| C: H&E of cornea with macular dystrophy. Note anterior stromal deposits and Bowman's layer disruption | D: Mucopolysaccharide deposits within keratocytes highlighted with Alcian Blue stain |
 |
 |
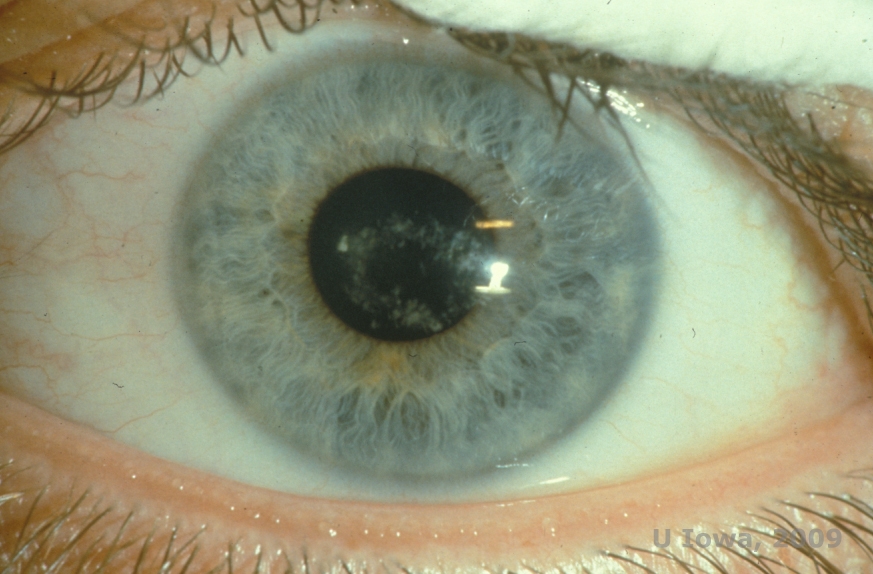
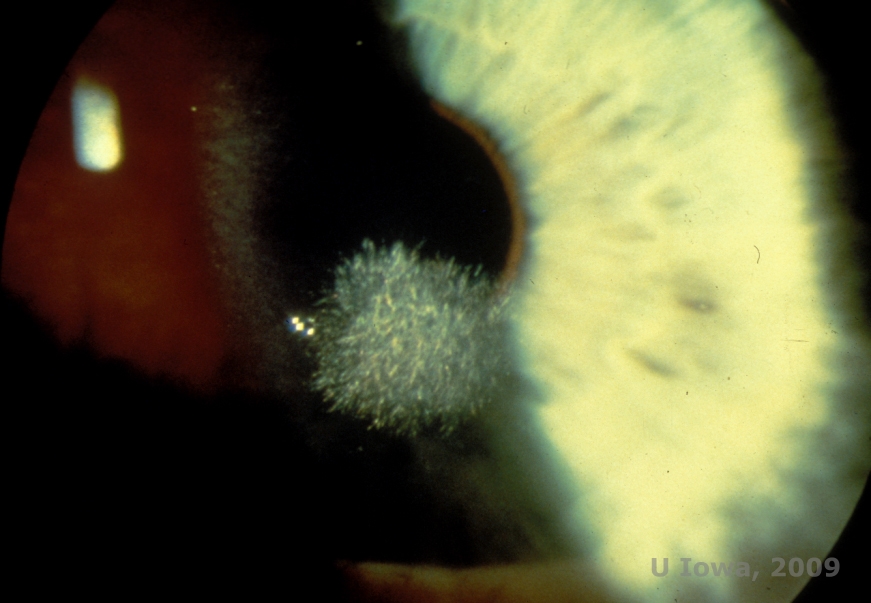
Schnyder corneal dystrophy (SCD), previously known as Schnyder crystalline corneal dystrophy, is an autosomal dominant, bilateral corneal stromal dystrophy linked to a genetic mutation in UbiA prenyltransferase domain containing 1(UBIAD1) gene on chromosome 1. The resulting metabolic defect of corneal keratocytes leads to crystalline cholesterol deposition in the stroma. However, presence of crystals is not absolutely necessary for the diagnosis of SCD. In fact, only 54% of patients with SCD have corneal crystals. Typically, patients present in the second or third decade with a ring-shaped central corneal opacity with or without comma-shaped subepithelial crystals (See Figure 6A and 6B). Then, arcus lipoides appears between the ages of 23 and 38. After age 38, progressive corneal clouding results in a panstromal haze reaching the midperiphery. Most patients over 50 years of age have photopic vision loss, glare, and decreased corneal sensation, and therefore, may require surgical treatment including corneal transplantation or PTK. Recurrence in the graft may occur. The disease has been linked with hypercholesterolemia, hyperlipidemia and genu valgum in some patients (2,5,9,10).
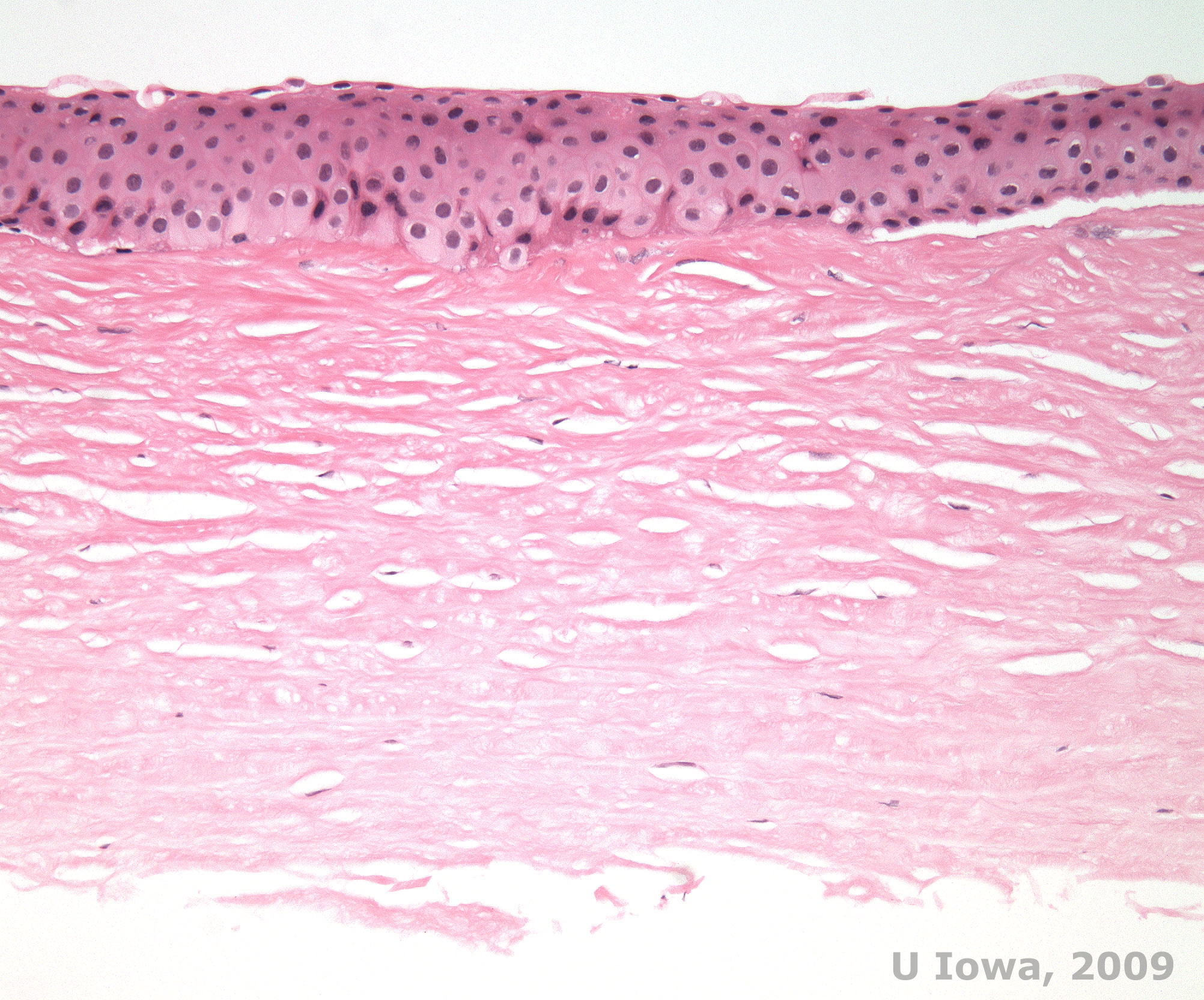
Histopathologically, birefringent cholesterol crystals composed of phospholipids and cholesterol deposit within basal epithelial cells, keratocytes, Bowman's layer, and between stromal lamellae. Lipids dissolve in normal histologic processing, so frozen sections through the cornea must be obtained to demonstrate the presence of lipid with Oil-Red-O or Sudan black stains.
| A: Slit lamp photo of Schnyder Corneal Dystrophy. | B. Centrally located crystalline deposits |
 |
 |
| C: H&E of cornea with SCCD | D. Oil Red O stain highlights cholesterol crystals which appear red. |
 |
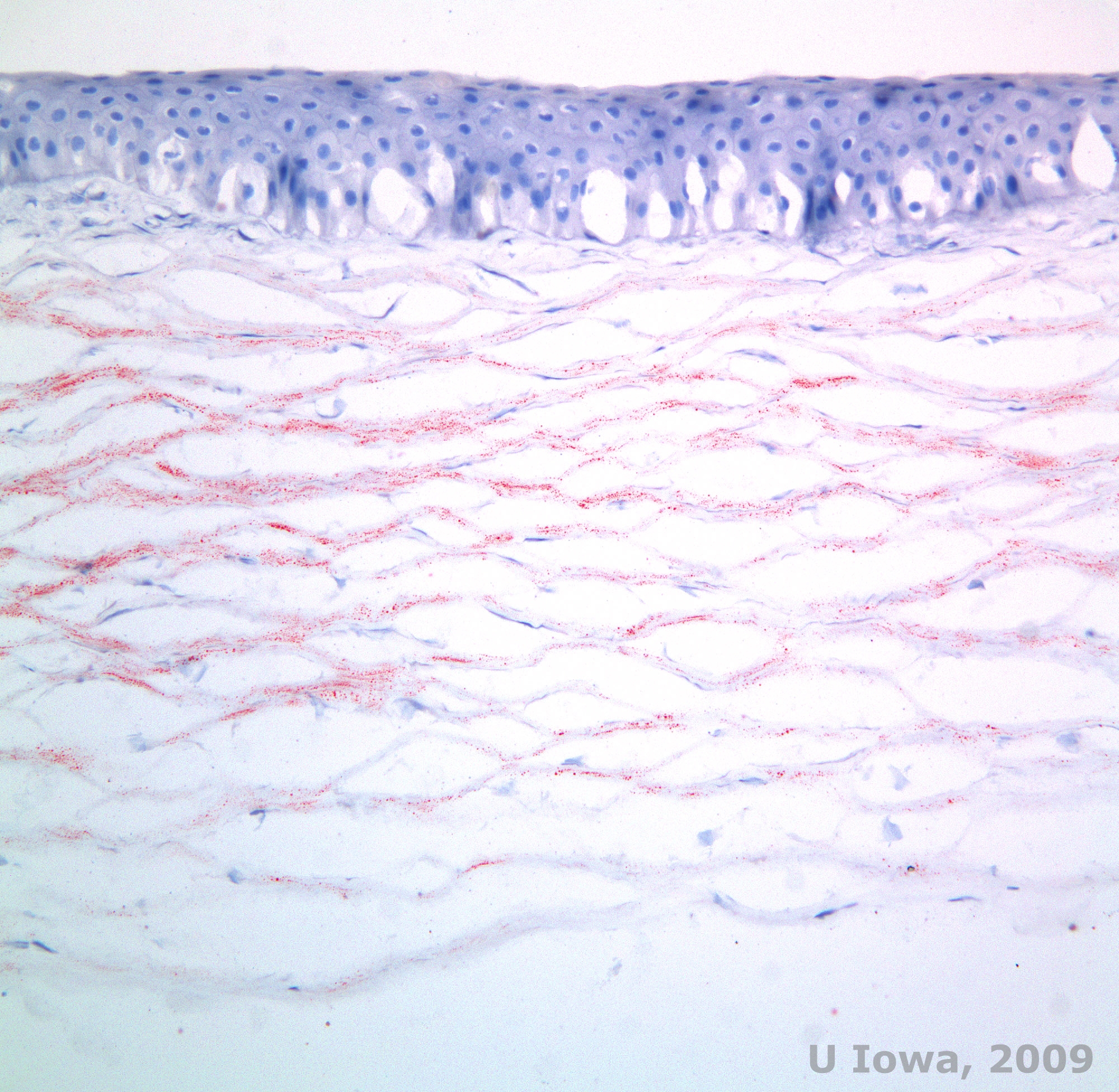 |
Table 4 provides a common mnemonic for memorizing some of the corneal dystrophies affecting the stroma, the composition of their deposit, and the method of staining these deposits is listed.
Table 4: Mnemonic for remembering corneal stromal dystrophies |
|
EPIDEMIOLOGY
|
SIGNS
|
SYMPTOMS
|
TREATMENT
|
Van C, Syed NA. Epithelial-Stromal and Stromal Corneal Dystrophies: A Clinicopathologic Review. Revision of [Birkholz ES, Syed NA, Wagoner, MD. Corneal Stromal Dystrophies: A Clinicopathologic Review. Aug. 17, 2009]; EyeRounds.org. August 20, 2015. Available from: http://www.eyerounds.org/cases/43-Corneal-Stromal-Dystrophies.htm

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
