
Chief Complaint: Referral for evaluation of Kayser-Fleischer Rings
History of Present Illness: The patient was a 25-year-old male who had been experiencing a right-sided subcostal tenderness and increasing fatigue for the last several months. He also had noticed dark urine, jaundice and a 60 pound weight loss. The patient was admitted for work-up and treatment of his undiagnosed condition. His inpatient team was concerned about possible Wilson's disease and referred the patient to the ophthalmology clinic for evaluation of Kayser-Fleischer rings. He had no changes in vision or other ocular complaints.
Of note, A liver biopsy at age 14, performed after he was found to have abnormal routine labs prior to a tonsillectomy, showed fatty liver which was thought to be related to his weight. He had no additional follow up.
Past Ocular History: None
Medical History: The patient had a liver biopsy at age 14 with a diagnosis of fatty liver. He also had a tonsillectomy at age 14.
Medications: None
Family History: There is no family history of liver disease. The patient's mother was an alcoholic.
Social History: The patient is currently incarcerated for burglary. He reports heavy alcohol use (more than one six-pack of beer per day for over eight years). He smokes one pack of cigarettes per day and reports prior use of marijuana, LSD, and cocaine in the past.
Review of Systems: The patient reported an unintentional 60 pound weight loss. He had no mood changes, behavioral changes, or movement disorders.
Ocular Exam:
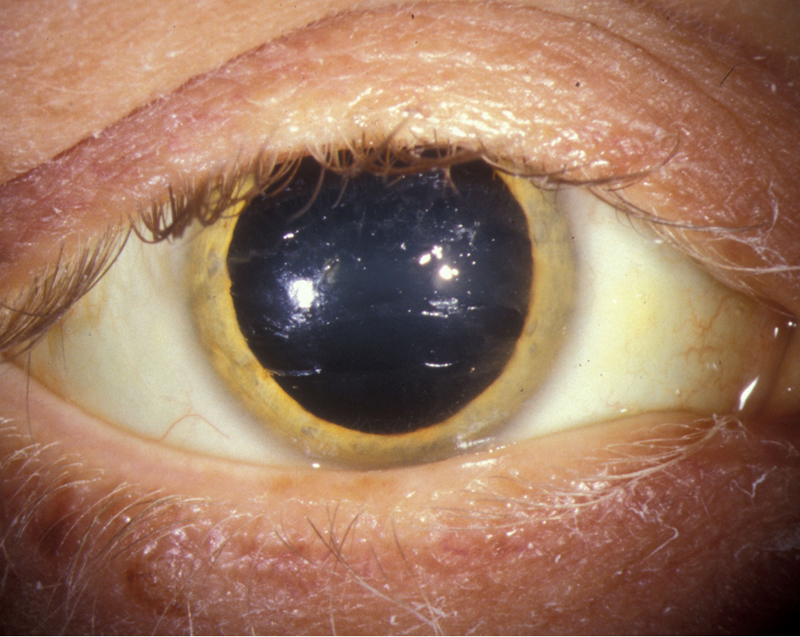
| Figure 1a: Golden Brown Kayser-Fleischer ring | Figure 1b: Higher magnification photo of golden brown ring at the level of Descemet's membrane |
 |
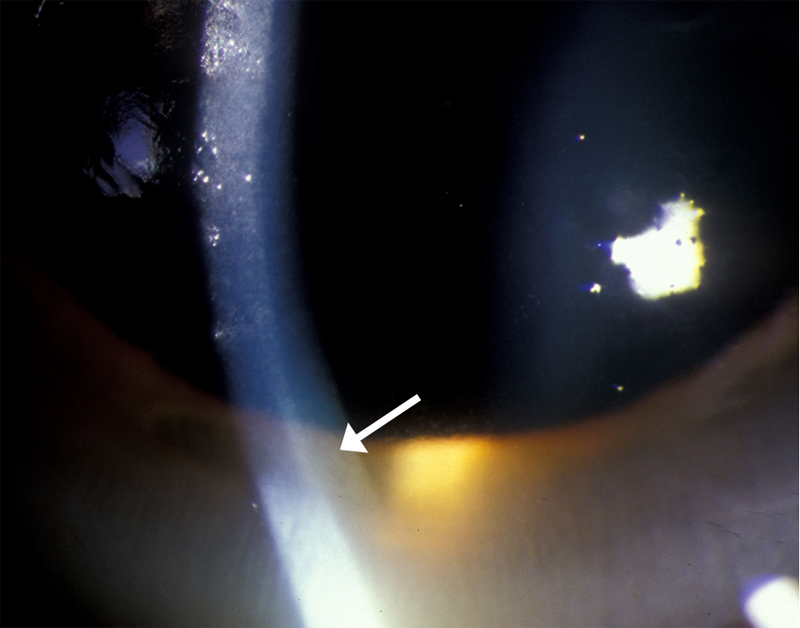 |
| Figure 1c: Gonioscopy of angle, showing golden brown deposit in Descemet's membrane | |
 |
|
The patient was admitted to the UIHC for a diagnostic work-up as well as treatment for severe liver failure. Subsequent work up revealed worsening liver function tests. He had normal serum ceruloplasmin and serum copper levels, but had an elevated urine copper of 189 ng/ml (normal is 12-80), and an elevated urine copper excretion of 1522 mcg/day (normal is 3-35) after penicillamine excretion test. His liver biopsy revealed a hepatic copper quantification of 763 mcg copper per gram of dry weight (normal is 10-35 mcg/gm).
Due to the presence of Kayser-Fleischer rings, elevated urine copper excretion after penicillamine excretion test, and diagnostic liver biopsy, the patient was diagnosed with Wilson's disease. He was then treated with penicillamine 250 mg 4 times a day and pyridoxine 25 mg PO daily which improved his urine copper clearance. However, he developed significant encephalopathy, a progressively worsening coagulopathy with hemolysis and anemia of liver disease, and significant ascites. His liver disease continued to progress over his two week hospital course, and he demonstrated a sub-fulminant course. His hepatologists felt the disease would likely progress to liver failure and likely death. Liver transplant was unable to be performed at UIHC due to his history of alcohol use and his poor support network. He was then transferred to a hospital closer to his family.
Wilson's disease, first described in 1912 by Kinnear Wilson, is a rare autosomal recessive disease of decreased biliary copper excretion and copper deposition throughout the body, most notably in the liver, brain, kidneys, and cornea. The disease is caused by a genetic mutation in the ATP7B gene on chromosome 13 which codes for a membrane-bound copper transporting ATPase found mostly in the liver (Tanzi, 1993). Patients with Wilson's disease can present with chronic liver disease, fulminant hepatic failure, acute renal failure, hemolytic anemia, or neuropsychiatric disease such as movement disorders, tremors, incoordination, and behavioral changes (See Table 1). The disease can present at any age, but is usually seen between the 1st and 4th decades of life (Schoen, 1990). The worldwide prevalence has been reported to be 1 in 30,000 (Scheinberg, 1984). Left untreated, this disease is lethal. Common treatments with penicillamine, trientine, and/or zinc therapy (copper chelation treatments) as well as low copper diets are life long endeavors, with liver transplantation being life-saving in very advanced cases (Mak, 2008).
| Hepatic | Neurologic | Psychiatric |
|---|---|---|
Jaundice Acute Hepatitis Cirrhosis Chronic Liver Disease Fulminant Hepatic Failure Acute Renal Failure Hemolytic Anemia |
Tremor Dystonia Bradykinesia Incoordination Insomnia Spasticity Chorea Drooling Seizures Migraine |
Schizophrenia Depression Manic Depressive Disorder Delusions Behavioral Disorders Personality changes |
Wilson's disease is usually suspected in young patients under the age of forty who have unexplained liver disease, unexplained neurological behavioral and/or psychiatric disease in the setting of liver disease, or a family history of Wilson's disease. The diagnosis of Wilson's disease is often based on Sternlieb's criteria, where a patient must have at least two of the following findings: the presence of Kayser-Fleischer rings, typical neurologic symptoms, and/or low ceruloplasmin levels (<0.20 g/L) (Sternlieb, 1990). Unfortunately, these criteria are often only met when a patient presents with advanced disease and usually have neurologic and/or psychiatric manifestations. Patients with early disease, hepatic disease only, or asymptomatic disease are very difficult to diagnose.
No laboratory test exists that reliably identifies Wilson's disease, but a combination of tests including serum ceruloplasmin, serum free copper, and 24 hour urinary copper excretion, are used together to identify abnormalities in copper metabolism.
Ceruloplasmin is a protein made by hepatocytes that binds copper and delivers it to peripheral tissues. When copper is unavailable, due to deficient transmembrane transport, to be incorporated into the apoceruloplasmin molecule as in Wilson's disease, the released apoceruloplasmin is rapidly metabolized, causing circulating levels of ceruloplasmin to be low. The normal level of ceruloplasmin in the blood is 0.20 to 0.40 g/L, however, this value is not applicable to pediatric patients, pregnant patients, or patients on estrogen. Other diseases can cause low levels including malnutrition, nephrotic syndrome, hereditary aceruloplasminemia, or inflammatory disorders (as it is an acute phase reactant). Also, ceruloplasmin levels may be normal in patients with Wilson's disease, quoted as anywhere from 35% -45% in patients with hepatic presentation and 60% of patients with fulminant hepatic failure (Steindl, 1997). Therefore, in patients with liver disease, a normal ceruloplasmin level cannot exclude Wilson's disease, nor is a low level sufficient to make a diagnosis of Wilson's disease.
|
24 hour urinary copper excretion is always elevated in patients with Wilson's disease (>100 µg/day or 1.0 mol/day), but accurate results are difficult to obtain as compliance may be low, incomplete collection may occur, and contamination from exogenous copper may occur as when the urine collection container is rinsed with tap water. Measuring the 24 hour urinary copper excretion before and after administration of D-penacillamine can help differentiate patient's with Wilson's from patients with other liver disorders as patients with Wilsons' will have excretion of more than 25 mol/day. This test has been shown to have a sensitivity of 76-88%% and specificity of 93-98% (Martins da Costa, 1992 and Muller, 2007). This test was not, however, reliable for diagnosing asymptomatic patients with Wilson's disease (sensitivity of 46%) and is quite cumbersome for patients to perform (Muller, 2007).
More recently, there have been reports of utilizing the alkaline phosphatase to total bilirubin ratio and/or the AST to ALT ratio to assist in the diagnosis of Wilson's disease in the setting of acute liver failure. A publication by Korman et al. reports that an alkaline phosphatase to total bilirubin ratio of less than 4 yielded 94% sensitivity and 96% specificity with a likelihood ratio of 23 for diagnosing fulminant Wilson's disease. They also report that an AST:ALT ratio of greater than 2.2 was 94% sensitive and 86% specific with a likelihood ratio of 7 for diagnosing fulminant Wilson's disease, and when the tests were combined, the diagnostic sensitivity and specificity was 100% (Korman, 2008).
The gold standard test for diagnosing Wilson's disease is measuring hepatic copper content on a liver biopsy. According to Ferenci et al. a copper content of >250 µg/g dry weight (normal, up to 50 µg/g dry weight) is 83% sensitive and 98.6% specific for diagnosing Wilson's disease, but performing liver biopsies is an invasive procedure and risky in patients with severe liver failure who have coagulopathy (Ferenci, 2005).
Genetic testing of the ATP7B gene can be performed with high sensitivity if the mutation is known from a proband or if the patient has one of the common genetic mutations for Wilson's disease. However, genetic testing is not routinely used as a diagnostic tool as testing is cumbersome to perform due to the gene's long length (21 exons), its numerous mutations (over 70 different mutations), and the fact that most patients have two different mutations (compound heterozygotes) (Mak, 2008).
Due to difficulty in diagnosing Wilson's disease, a scoring system was created and promoted by the 8th International meeting on Wilson's disease which is based on seven criteria, including the presence of Kayser-Flesicher rings; typical neurological symptoms; decreased serum ceruloplasmin concentration; Coombs' negative hemolytic anemia; elevated urinary copper excretion; high liver copper value in the absence of cholestasis; and mutational findings. Like all other laboratory testing, this scoring system tends to be more reliable in patients with advanced disease (Ferenci, 2003).
The Kayser-Fleischer ring is the hallmark of Wilson's disease and its detection may be critical for diagnosis. There are reports where it has been the first detectable manifestation of Wilson's disease, which led to early diagnosis and treatment for the disease (Liu, 2002).
The presence of Kayser-Fleischer rings in combination with low serum ceruloplasmin is considered diagnostic of Wilson's disease based on Sternlieb's criteria (Martins da Costa, 1992). In the cornea, the excess circulating copper is deposited in Descemet's membrane and is usually seen as a golden brown ring located in the peripheral cornea, beginning at Schwalbe's line and extending less than 5 mm onto the cornea (see video). The ring may also appear as greenish yellow, ruby red, bright green, or ultramarine blue. It is almost always bilateral and appears superiorly first, then inferiorly, and then later becomes circumferential (Kim, 1979). In the earlier stages of disease, gonioscopy is often needed to detect this subtle finding, but in advanced disease it can be seen with the naked eye.
|
Video 1: Golden Brown Kayser Fleischer ring |
These rings have been reported to be seen in about 85%-100% of patients with neurological and/or psychiatric manifestations of Wilson's disease but only 33%-86% of patients with hepatic disease and 0%-59% of asymptomatic patients (Mak, 2008). Kayser-Fleischer rings may be absent in up to 50% of patients with Wilsonian liver disease and in an even higher proportion with fulminant Wilsonian liver disease (Steindl, 1997). There are a number of conditions that have also been linked to colored rings in the cornea, including other liver diseases such as primary biliary cirrhosis, neonatal hepatitis, and cryptogenic cirrhosis, or elevated copper for other reasons such as in multiple myeloma, pulmonary carcinoma, benign monoclonal gammopathies, chronic lymphocytic leukemia, or even oral contraceptive use. After the initiation of treatment, the Kayser-Fleischer ring disappears in 85-90% of cases (Lossner, 1986).
At the University of Iowa, the ophthalmology on-call clinic and inpatient service frequently receive consults to evaluate for Kayser-Fleischer rings in patients with unexplained liver disease, but few ophthalmologists have ever seen a true Kayser Fleischer ring. In a survey performed of all attendings, fellows, and residents at UIHC, only 11 physicians reported ever seeing a Kayser-Fleischer ring (41%), with a total of reported cases being 24 over the entire career of all the ophthalmologists surveyed. Often, the Kayser-Fleischer rings of one patient were seen by multiple ophthalmologists in the department so the total number of patients diagnosed is less than the total number of reported cases seen. Of those that had seen a Kayser-Fleischer ring, only six ophthalmologists had seen more than one case. For 64% of the physicians, the identification of a Kayser-Fleischer ring was helpful in the diagnosis of Wilson's disease in at least one case.
A survey of six hepatologists at the University of Iowa, a tertiary care center, revealed only four new diagnoses of Wilson's disease over a mean clinical practice period of 13 years per physician. According to the survey, none of the reported cases had a Kayser-Fleischer ring.
The neurology service was also surveyed regarding their experience in diagnosing Wilson's disease. One neurologist, who has practiced for 37 years has seen only one patient with Wilson's disease, and the patient had a Kayser-Fleischer ring. One other neurologist who participated in the survey has practiced for seven years and seen no patients with Wilson's disease.
With the ever-rising cost of healthcare in mind, an investigation into the average cost of diagnosing a patient with Wilson's disease at UIHC was performed. Specialty consultation as well as laboratory investigation is required to make the diagnosis in most cases. For the basic laboratory evaluation alone, the cost is $190 (AST, ALT, Alkaline phosphatase, Total Bilirubin, and serum ceruloplasmin), not including the other laboratory tests that would normally be drawn in a patient with any liver disease, including hemoglobin, platelets, basic metabolic panel, and albumin levels (See Table 3a). In cases of diagnostic difficulty (early presentation or hepatic disease only), the diagnosis may cost up to $605 for laboratory evaluation (including liver function tests, ceruloplasmin, and 24-hour urinary copper excretion), in addition to outpatient consultation fees for ophthalmology ($300), hepatology ($935), and/or neurology ($775), for a total cost of $2,615 (See Tables 3a, 3b, and 3c). The total cost for performing the gold standard liver biopsy is $3,105, which includes the physician fee, hospital fee, pathology fee as well as the send out laboratory value for hepatic copper content. In total, if all laboratory tests, consultation services, and liver biopsy were utilized, the total cost would be $5,720 (See Table 3c
| Lab Test | Cost |
|---|---|
| AST | $29 |
| ALT | $34 |
| ALP | $32 |
| Total Bilirubin | $29 |
| Serum Ceruloplasmin | $66 |
| 24-h urine copper excretion | $70 |
| Hepatic Copper Content (from liver biopsy send out lab) |
$275 |
| Consult/Procedure | Cost |
|---|---|
| Ophthalmology outpatient consult (low-moderate complexity) | $300 |
| Ophthalmology inpatient consult (low-moderate complexity) | $280 |
| Neurology Outpatient consult (moderate-high complexity) | $775 |
| Neurology Inpatient Consultation (moderate-high complexity) | $650 |
| Hepatology Outpatient Consultation (moderate-high complexity) | $935 |
| Hepatology Inpatient Consultation (moderate-high complexity) | $860 |
| Liver Biopsy: Total (outpatient costs) | $2,830 |
| Physician charge | $1,226 |
| Hospital charge | $926 |
| Pathology professional fee | $312 |
| Pathology technical fee | $366 |
| services | cost |
|---|---|
Diagnosis without ophthalmology consultation |
$2,315 |
Diagnosis with ophthalmology consultation |
$2,615 |
Total cost of liver biopsy |
$3,105 |
Diagnosis with all common tests, consults, and liver biopsy |
$5,720 |
Although extremely rare at UIHC with only four to five new diagnoses of Wilson's disease in an average time period of 13 years based on a survey of hepatologists, an ophthalmologic evaluation to look for Kayser-Fleischer rings is still a very essential diagnostic tool and is a non-invasive, affordable way to assist in the diagnosis of a potentially fatal disease. However, few ophthalmologists are experienced in identifying a Kayser-Fleischer ring, as only 41% of the ophthalmologists surveyed at the University of Iowa have ever seen one.
When a patient presents with advanced disease or neurological and/or psychiatric manifestations of Wilson's disease, a Kayser-Fleischer ring is present in almost all cases and can non-invasively help to solidify the diagnosis. When a patient presents with less advanced disease or hepatic disease only, the diagnosis is much more difficult and the critical evaluation of all available tests is often needed to confirm the diagnosis. Because many laboratory tests are inconclusive in patients with less advanced disease, and because the gold standard liver biopsy is an invasive and high cost procedure, the non-invasive evaluation for a Kayser-Fleischer ring is still an essential part of the diagnostic work up for Wilson's disease. In these cases identifying a Kayser-Fleischer ring is often much more difficult as the ring may be very faint and identifiable only on gonioscopy. Therefore, an evaluation in the clinic utilizing a slit lamp and gonioscopy lens is essential in this group of patients.
Special thanks to Dr. Michael Voigt, Dr. Bruce Luxon, Dr. Stephanie Dee, Dr. Kyle Brown, Dr. Douglas LaBrecque, and Dr. Warren Schmidt , all part of the Hepatology division of the Gastroenterology department at The University of Iowa Hospitals and Clinics (UIHC). Thanks also to Dr. Robert Rodnitzky and Dr. Pedro Gonzalez in the department of Neurology at UIHC.
Birkholz ES, Oetting TA. Kayser Fleischer Ring: A Systems Based Review of the Ophthalmologist's Role in the Diagnosis of Wilson's Disease. EyeRounds.org. July 28, 2009; Available from: http://www.EyeRounds.org/cases/97-kayser-fleischer-ring-Wilsons-Disease.htm.

Ophthalmic Atlas Images by EyeRounds.org, The University of Iowa are licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License.

Address
University of IowaLegal
Related Links
