Session I - Introduction by Session Chair Stewart Thompson, Ph.D.
Session II - Introduction by Session Chair Akihiro Ikeda, Ph.D.
Session III - Introduction by Session Chair D.J. Sidjanin, Ph.D.
Poster Presentations
Abstracts
The Role of Rabac1 in Photoreceptor Development
Ameair Abu-Irqeba, Judith Ogilvie
Biology Department, Saint Louis University, St. Louis, MO
Purpose: The Rd1 mouse retina was the first identified animal model of the human disease retinitis pigmentosa (RP), characterized by early-onset, rapid degeneration of rod photoreceptors between postnatal days 10-21 (P10-21). The Rd1 rods show developmental defects significantly before degenerative changes appear, including failure to undergo normal synaptogenesis. In preliminary studies, we have performed microarray analysis of wild type mouse retina compared to Rd1 mutant retinas at P2-8, prior to onset of photoreceptor death. In addition to the mutant gene, one other gene was confirmed to be down-regulated at all time points examined - Prenylated Rab Acceptor 1 (rabac1), which codes for the protein PRA1. This protein is important for vesicular trafficking and we predict that it is important for normal rod development as well as protein trafficking to the outer segment.
Methods: RNAi and overexpression constucts were cloned into vectors that contain a CAG promoter. They were then tested for efficacy in a mammalian cell line prior to subretinal injection (and electroporation) into the retina. The RNAi constuct was delivered into WT mouse retina. Rabac1-GFP was delivered into the Rd1 mouse retina.
Results: No difference was identified between WT photoreceptors that receive the PRA1 RNAi vector and those that did not. Over-expression of Rabac1 in Rd1 photoreceptors did not lead to a rescue of the degeneration phenotype.
Conclusions: Over-expression of PRA1, which is normally downregulated in Rd1 retina, does not lead to rescue of the photoreceptor degeneration phenotype. Knockdown of PRA1 in WT retina does not lead to degeneration. This may mean that PRA1 could have other developmentally important functions that are not essential for survival of photoreceptors.
Back to top
Investigation of Gene-Expression Patterns in Familial Angle-Closure Glaucoma in the Basset Hound
Dina Ahram1, Sinisa D. Grozdanic2, Helga Kecova2, Markus H. Kuehn1
1Interdisciplinary Graduate Program in Genetics, University of Iowa, Iowa City, IA; 2Department of Veterans Affairs � Center for Prevention and Treatment of Vision Loss, Iowa City
Purpose: Primary angle closure glaucoma (PACG) is a condition that most commonly results from the collapse of the irido-corneal angle due to the anterior movement of the iris. We have identified several Basset Hound pedigrees with characteristic autosomal recessive PACG that closely recapitulates the clinical PACG phenotype observed in human patients and examined the inherent variation in gene-expression patterns in affected and unaffected animals. Gene expression assessment of cultured scleral cells was performed in order to identify genes with potentially significant down-regulation or abolished expression without the background effects of inflammatory activity typically found in the glaucomatous eye.
Methods: A primary fibroblast cell culture was established from the sclera of three PACG and three unaffected Bassets. Total RNA extracted from fibroblast cells was assayed using the Affymetrix GeneChip Canine Genome 2.0 Array. The Robust Multichip Average expression summary method was used for background adjustment and normalization. A two class, unpaired, Wilcoxon statistical test was conducted to identify differentially expressed genes. qRT-PCR was performed to validate significantly expressed genes.
Results: PACG fibroblast cultures were observed to display slow growth rates and dysmorphic cell appearance in comparison to wild-type cells. Gene expression analysis revealed over 736 differentially expressed genes based on a minimum two-fold change cutoff in expression level. Genes which revealed significant expression down-regulation in PACG versus control cells include EGFLAM, PSAT1, DRAM1, RASGEF1B, GOLGA1, PCK2 and FAP. Validation of differentially expressed genes using qRT-PCR revealed a significant fold change in genomic DNA quantity in PACG versus control cells.
Conclusions: These studies further suggest that cellular dysfunction is an important aspect in the pathophysiology of PACG in the dog. The identification of genes with significantly altered expression levels does not only provide insight into the molecular pathways associated with the development of PACG but will be helpful in the future characterization of the genetic defect underlying the disease. Ultimately, we anticipate for these studies to also provide valuable insight into the pathophysiology and genetics of human PACG
Back to top
Toward a mechanism linking Glaucoma and Corneal thickness: Candidate gene screening for Cctql
Maurisa Aimable1, Demelza Koehn1, Michael G. Anderson2
Departments of 1Molecular Physiology & Biophysics, 2Ophthalmology & Visual Sciences, The University of Iowa, Iowa City, Iowa
Purpose: Glaucoma is one of the leading causes of blindness in the world affecting around 70 million people worldwide. Central corneal thickness (CCT) has become recognized as a major risk factor. The biological reasons behind this association remain largely unknown. In order to study the associations between corneal thickness and glaucoma, we have undertaken a phenotype-drive approach using inbred strains of mice. We have identified a region on mouse chromosome 7 that plays a significant role in determining differences in corneal thickness between two inbred strains of mice, C57BLKS/J and SJL/J. The purpose of this study was to seek the causative variation(s) within the chromosome 7 critical region by sequencing candidates within the interval and establishing functional tests for any putative mutations identified.
Methods: Candidate genes were selected based on known corneal gene expression, differential expression between inbred KS and SJL mice, and gene function. Candidate genes were analyzed by sequence analysis to identify any significant changes.
Results: Based on our selection criteria, five genes were selected. Thus far three of the five have been analyzed. One gene did not contain any significant differences in the SJL or KS strain while another contained no changes in the KS strain and five synonymous changes in the SJL strain. The remaining gene contained two synonymous changes, twelve intronic changes and one non-synonymous base pair substitution.
Conclusions: Our goal is to find a defect in one or more of our candidate genes in mice and be able to understand the defect in the molecular pathway. Our current experiments have found genetic changes, but not that are definitively mutations. In ongoing work, we are establishing biological tests for the most promising variation we have found and increasing our throughput of ongoing sequencing analyses.
Back to top
Use of Optical Coherence Tomography (OCT) in mouse sub-retinal gene therapy trials
S. Bhattarai, D. Gratie, M. Stunkel, R.F. Mullins, A.V. Drack
The University of Iowa Department of Ophthalmology and Visual Science, Iowa City, IA
Purpose: Subretinal injection of viral vectors is used to treat retinal degeneration in mouse models. Traumatic complications may occur. We used OCT to screen mouse eyes after injection to determine how many eyes develop traumatic changes from the injection.
Methods: Mice received subretinal injection of 2 microliters AAVBbs1FLAG or AAVGFP. Using an operating microscope, a temporal conjunctival peritomy was made, followed by a stab incision using a sharp 30 gauge needle, then injection into the subretinal space using a 33 gauge blunt needle on a Hamilton syringe. 1-2 weeks after injection, posterior segment Bioptigen OCT was performed using a volumetric scan with 400 horizontal and 400 vertical scans inside a 1mm 2 to 1.4 mm2 area. The first scan was centered on the optic nerve, then the probe was reoriented to scan peripherally. Histology was performed.
Results: 42 eyes of 22 mice were injected then screened with OCT. 9/42 eyes (21%) had a chronic retinal detachment. 8/42 (19%) had other signs of trauma. 6/42 had an identifiable needle entry site without any other signs of trauma. 19/42 had no identifiable needle track or trauma. 60% of eyes had no reason for exclusion.
Conclusion: A significant proportion of subretinally injected eyes have traumatic damage that may skew results if not recognized. OCT is a rapid and reliable means of determining early which eyes should be excluded. In addition, the ability to identify the needle track on OCT can assist with localization of transduced retina.
Back to top
More Numerous Photosensitive Ganglion Cells In A Macular Degeneration
Frederick R. Blodi1, Steven F. Stasheff1,2, Stewart Thompson2, Malini Shankar1, Robert F. Mullins2, Michael P. Andrews1, Edwin M. Stone2
Departments of 1Pediatrics, 2Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, IA
Purpose: We sought to determine the origin of increased behavioral sensitivity to light in a mouse model of the inherited early-onset macular degenerative disease malattia leventinese (Efemp1R345W). We previouslyfound that this mutation caused an unexpected increase in sensitivity to light for a behavior (negative masking) mediated by intrinsically photosensitive retinal ganglion cells (ipRGCs). Next, we determined that Efemp1 localizes to the retinal ganglion cell layer, and hypothesized that mutant Efemp1 was altering the light responsiveness of retinal ganglion cells. We now report an increased number of ipRGC-like responses in Efemp1R345W mice.
Methods: We used in vitro multielectrode recording to monitor retinal ganglion cell activity in wild type (wt) and Efemp1R345W mice. Action potentials were recorded as full field broadband light flashes were presented (peak irradiance 1016-1018 photons/cm2·s at 548 nm, duration 2- 25 sec) to retinas perfused with control Ringer’s solution and one designed to pharmacologically prevent all synaptic transmission, thus revealing those cells with intrinsic photosensitivity.
Results: In wt retinas under pharmacologic blockade of synaptic transmission, typically 3-4 ganglion cells per retina were detected that had responses characteristic of ipRGCs (relatively long latency and duration). In contrast, 15-25 such cells were recorded in retinas from Efemp1R345W mice.
Conclusions: Our recordings indicate that Efemp1R345W mutations can lead to an increase in the number of ganglion cells that exhibit intrinsic responses to light. This might be due to an overall greater number of melanopsin-containing ipRGCs in mutant retinas relative to wt. Alternatively, there may be increased signal propagation from ipRGCs to other classes of ganglion cells not normally directly responsive to light. In either case, this identifies a novel function for Efemp1 in the retina.
Back to top
Optic nerve axon loss in a Cat Model of Inherited Glaucoma : validation of a semi-automated targeted sampling method
Owen Bowie3, Leandro B. Teixeira1,2, T M. Nork3,4, Richard R. Dubielzig1,4, Gillian J. McLellan3,4
1Pathobiological Science, UW-Madison School of Veterinary Medicine, Madison, WI; 2Comparative Ophthalmic Research Laboratories, Madison, WI; 3Ophthalmology and Visual Sciences, UW-Madison, Madison, WI; 4University of Wisconsin Eye Research Institute, Madison, WI.
Purpose: To validate a semi-automated method for quantifying axons in the optic nerves of glaucomatous cats.
Methods: Single optic nerves were selected from cats with mild to severe glaucoma (n= 9) and normal cats (n=6). Following perfusion fixation and enucleation, samples of optic nerve were dissected,,fixed in glutaraldehyde, osmicated and resin embedded. Semi-thin sections stained with 1% p-phenylenediamine were evaluated by light microscopy. Axons were quantified using both full-count, and semi-automated targeted sampling (SATS) methods. Full counts were obtained from up to 40 non-overlapping photomicrographs encompassing the entire nerve cross-section. Up to 3 distinct, relatively homogeneous regions were identified. An automated counting function was used to calculate the average axon density, from up to 5 randomly selected photomicrographs for each region. An estimate of the total number of axons in each cross-section was thus obtained. Three masked observers obtained counts on the same sections by the SATS method to evaluate inter-observer reproducibility.
Results: Median total axon count (full count method) was significantly lower in the glaucomatous cats (39020) than normal cats (62713; p=0.0016, Mann-Whitney). Correlation between the semi-automated counts and total counts was strong (linear regression slope = 1.10, y-intercept =564 and r2=0.96) with acceptable inter-observer reproducibility.
Conclusions: The SATS method provides a practical, rapid and relatively reliable means of quantifying optic nerve axon numbers in normal cats and cats with glaucoma. Evaluation of the relationships between optic nerve damage and in vivo measurements of optic nerve structure and function in this spontaneous animal model of glaucoma is ongoing.
Back to top
Endothelial Progenitor Cell Response to Oxygen Induced Retinopathy
B.E. O’Bryhim1,2, R.S. White1, R.C.A. Symons1,2
University of Kansas Medical Center, 1Department of Ophthalmology, 2Department of Molecular and Integrative Physiology
Purpose: Bone marrow-derived endothelial progenitor cells (EPCs) have been shown to contribute to angiogenesis in a variety of pathologies, including metastatic disease, wound healing, and diabetic vascular disorders. Recent work has indicated that EPCs additionally play an important role in both retinopathy of prematurity (ROP) and in the murine model of ROP, oxygen-induced retinopathy (OIR). The purpose of this study is to characterize response of this cell population to OIR during vaso-obliteration and revascularization.
Methods: C57BL/6J pups were exposed to 75% oxygen for 120 hours beginning on post-natal day 7 (P7). Mice were sacrificed at daily time points from P7 to P13, and also at P7.5 and P9.5. Retinas were stained for the presence of CD34+/AC133+ EPCs. Live, CD45-/CD34+/AC133+ EPCs were enumerated in bone marrow and blood samples using flow cytometry.
Results: The number of EPCs fluctuated early after exposure to OIR, but remained constant from P10 through P13. The data indicated an initial decrease in the number of EPCs in the bone marrow in response to hyperoxia, with a peak increase on P9 followed by a gradual decline. The number of circulating EPCs increased early in OIR but decreased from P8 to P10, after which the number increased through P12.
Conclusions: Bone-marrow derived EPCs are a dynamic population of cells that respond to tissue injury caused by extended hyperoxic stress. A better understanding of their contribution to vascular damage and repair may serve to improve therapies available for vasoproliferative retinopathies, including ROP.
Back to top
 Voted Outstanding Poster Presentation
Voted Outstanding Poster Presentation
LCA Gene Therapy In Somatic-Cell-Derived Induced Pluripotent Stem Cells
Erin R. Burnight, Emily Kaalberg, Simon Petersen-Jones, Robert F. Mullins, Edwin M. Stone, Budd A. Tucker
Institute for Vision Research, The University of Iowa, Iowa City, IA
Purpose: The purpose of this study is to generate induced pluripotent stem cells (iPSCs) and subsequently photoreceptor precursor cells from animal models of and patients with the CEP290-associated Leber Congenital Amaurosis (LCA). These cells will be used for the study of therapeutic gene correction.
Methods: Fibroblast-derived iPSCs were generated from animal models and an LCA patient with molecularly confirmed CEP290 mutations using viral reprogramming vectors. iPSCs were examined for the presence of pluripotency marker transcripts and proteins. Animal model and human iPSCs were differentiated into photoreceptor precursors using our previously developed step-wise differentiation protocol. HIV-1 lentiviral vectors were developed to generate reagents for cell-specific therapeutic transgene expression.
Results: rt-PCR and immunocytochemical analysis of iPSCs and differentiated cells confirmed pluripotency and photoreceptor marker expression, respectively. The human CEP290 cDNA was packaged into a lentiviral vector. Vector-derived CEP290 mRNA expression was detected in JK1 cells. We observed cell death following transduction with high doses of HIV-CEP290 indicating that overexpression of CEP290 is toxic. Lentiviral vectors expressing GFP from retina-specific promoters were tested in differentiating primary retina culture. CRX-, NRL-, and RhoK – driven GFP expression was detected in a time-dependent manner.
Conclusions: We show successful somatic cell reprogramming and differentiation of animal model and human CEP290-LCA iPSCs into photoreceptor precursors. Lentiviral vectors expressing human CEP290 from retina-specific promoters have been generated and will be used in subsequent gene replacement therapies and transplantation studies. This work will contribute to our overall goal of vision restoration in patients with LCA through gene and cell therapy using patient-specific stem cells.
Back to top
Validation of Tablet-based Evaluation of Color Fundus Images
Mark Christopher1, Daniela C. Moga2, Stephen R. Russell3, James C. Folk3, Todd E. Scheetz1,3, Michael D. Abramoff3,4,5
University of Iowa Departments of 1Biomedical Engineering, 2Epidemiology, 3Ophthalmology and Visual Sciences, 4Electrical and Computer Engineering, 5Department of Veterans' Affairs, Iowa City, IA
Purpose: To compare diabetic retinopathy (DR) referral recommendations made by viewing fundus images using a tablet computer with those made using a standard desktop display.
Methods: A tablet computer (iPad) and a desktop computer with a high-definition color display were compared. For each platform, 2 retinal specialists independently rated 1,200 color fundus images from patients at risk for DR using an annotation program Truthseeker. The specialists determined whether each image had referable DR and also how urgently each patient should be referred for medical examination. Graders viewed and rated the randomly presented images independently and were masked to their ratings on the alternative platform. Tablet-based and desktop display-based referral ratings were compared using cross-platform intraobserver kappa as the primary outcome measure. Additionally, interobserver kappa, sensitivity, specificity, and area under the receiver operating characteristic were determined.
Results: A high level of cross-platform intraobserver agreement was found for the DR referral ratings between the platforms (κ = 0.778) and for the 2 graders (κ = 0.812). Interobserver agreement was similar for the 2 platforms (κ = 0.544 and κ = 0.625 for tablet and desktop, respectively). The tablet-based ratings achieved a sensitivity of 0.848, a specificity of 0.987, and an area under the receiver operating characteristic of 0.950 compared with desktop display-based ratings.
Conclusions: In this pilot study, tablet-based rating of color fundus images for subjects at risk for DR was consistent with desktop display-based rating. These results indicate that tablet computers can be reliably used for clinical evaluation of fundus images for DR.
Back to top
Pathogenic or Protective: Understanding the role of interleukin-17A in a spontaneous model of autoimmune uveitis elicited by retina-specific T cells
Benjamin C. Chaon1,2A,3, Reiko Horai2A, Jun Chen2A, Carlos Zárate-Bladés2A, Chi-Chao Chan2B, and Rachel R. Caspi2A
1. Howard Hughes Medical Institute – National Institutes of Health Research Scholars Program Bethesda, MD 2. Laboratory of Immunology: AImmunoregulation Section BImmunopathology Section National Eye Institute, NIH, Bethesda, MD 3. University of Iowa Roy J. and Lucille A. Carver College of Medicine Iowa City, IA
Purpose: Th17 cells and their cytokine product, IL-17A are implicated as major pathogenic mediators in autoimmune disease models induced by immunization with self-antigen, including uveitis. Recently, we generated transgenic (R161H) mice that express a T cell receptor specific to the retinal protein IRBP and spontaneously develop uveitis by two months of age. This study aims to examine the role of Th17 cells and IL-17A in the pathogenesis of uveitis in this spontaneous model.
Methods: R161H mice were crossed to IL-17A KO mice (R161H-17KO) or to IL-17A-GFP reporter mice (R161H-17Agfp). Disease was monitored by fundoscopy and confirmed by histology.
Results: IL-17A-GFP+ cells were detected exvivo in the uveitic eyes and lymph nodes of R161H-17Agfp mice. The majority of ocular infiltrating IL-17A-GFP+ cells were CD4+, exhibited a memory (CD62LloCD44hi) phenotype, and expressed the Th17-associated chemokine receptor, CCR6. R161H-17KO mice showed a slower disease onset, but no reduction in final disease scores compared to their R161H IL-17A sufficient littermates. A compensatory increase in IRBP-specific IL-17F, IFN-g,and GM-CSF production was observed in lymphocytes from R161H-17KO mice compared to IL-17A-sufficient controls. Interestingly, adoptive transfer of Th17- polarized, IRBP-specific lymphocytes with genetic deficiency of IL-17A, so-called “Th17 wannabes,” transferred disease of similar severity to that of mice receiving IL-17A sufficient, Th17-polarized, IRBP specific cells.
Conclusion: IL-17A is produced by IRBP-specific T cells and may contribute to inflammation – particularly during early stages of disease. Nevertheless, IL-17A appears to be dispensable in this model as compensatory increases in other inflammatory cytokines may be sufficient to drive disease in its absence.
Back to top
Utilzing The Lystbg-J Mutation To Identify Genetic Pathways Of Exfoliation Syndrome And Circadian Intraocular Pressure
Tryphena L. Cuffy1, Colleen M. McDowell2, Michael G. Anderson1,2,3
1Interdisciplinary Graduate Program in Genetics, 2Molecular Physiology and Biophysics, 3Ophthalmology and Visual Sciences, University of Iowa, Iowa City, IA.
Purpose: Exfoliation syndrome (XFS) is a common age-related disorder characterized by the pathological accumulations of fibrillar exfoliative material in the anterior chamber of the eye. Patients with XFS can go on to develop exfoliative glaucoma; potentially as a result of an accumulation of exfoliative material at the drainage structures of the eye. This can lead to abnormal elevated regulation of intraocular pressure (IOP). Human eyes with XFS exhibit a striking pattern of Marcel-like iris transillumination defects. That pattern is recapitulated in mice containing the Lystbg-J mutation. Others have identified a small number of proteins capable of interacting with the LYST WD40 motif – one of these being CSNK2B. Testing CSNK2B, previous GST-pulldown experiments confirmed that wild-type LYST can bind to CSNK2B whereas LYSTbg-J can not. This result suggests that LYST may play a role in regulating activity or localization of CSNK2B
Methods: Biochemical assays were used to test whether CSNK2B substrates exhibit functional deficits in the presence of the Lystbg-J mutation. A rebound tonometer was used to measure intraocular pressure in wildtype and mice carrying the Lystbg-J mutation.
Results: Intraocular pressure measurements showed that C57BL/6J mice have a biphasic IOP pattern in which IOP was low during the light phase (13.3±2.4 mmHg) and high during the dark phase (15.1±1.9 mmHg). In contrast, mice with the bg-J mutation showed a constant intraocular pressure during the light cycle (12.7±2.5mmHg) with no significant rise in IOP during the dark cycle (12.4±1.8 mmHg). Experiments with two IOP reducing pharmacological drugs indicated that dysfunction of circadian IOP in B6-Lystbg-J mice is dependent on aqueous humor production.
Conclusions: Combined, these results implicate CSNK2B in the pathogenesis of XFS and a genetic pathway impacting circadian IOP regulation. Deciphering the pathogenesis of XFS would therefore be therapeutically beneficial and could lead to better treatment and management of the disease.
Back to top
RIDE – Repetitive element Insertion Detector for Exomes
Adam P. DeLuca1, S. Scott Whitmore1, Edwin M. Stone1,2, Todd E. Scheetz1, Terry A. Braun1
1The University of Iowa, Iowa City IA 2 Howard Hughes Medical Institute, Iowa City IA
Purpose: MAK was identified as a gene causing retinitis pigmentosa when exome sequencing of an affected patient revealed an insertion of an Alu element into an exon. Interestingly, the insert itself was not detected, but rather, nearby erroneous calls lead to the evaluation of the region by Sanger sequencing. We present here a novel and efficient computational method to detect insertions of repetitive sequence in exome sequencing data.
Methods: There are characteristic anomalies in the sequence alignment of the reads surrounding the sites of repetitive element insertions that can be used to detect the event. To test the performance of the detector, an in silico simulation was performed. An alternate reference genome was defined where annotated Alu elements were removed. Specifically, Alu elements located in regions captured in exome sequencing experiments were excised from the human reference genome (hg19/ GRCh37). Exome sequence data was then aligned to this modified reference genome. This simulates homozygous insertions like the one that lead to the discovery of MAK.
Results: RIDE has been implemented on top of the GATK framework to allow easy integration into existing workflows. Simulation results show a sensitivity of 89.1% with a false discovery rate of 16.1%.
Conclusion: RIDE closes an analytical hole in the processing of exome sequence data that contributes to the overall false negative rate of these experiments. This will enable the discovery of additional disease genes, potentially from previously sequenced samples where no causative mutation could be found.
Back to top
 Voted Outstanding Oral Presentation
Voted Outstanding Oral Presentation
Complement regulation in Mfrp174delG mice, a model for advanced dry AMD
Joseph Fogerty, Joseph C. Besharse
Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, WI
Purpose: We have characterized a naturally-occurring mutation in mouse that causes slow, progressive photoreceptor degeneration, white fundus flecks, and late-onset RPE atrophy. Genetic studies identified a deletion in the 5’ coding sequence of MFRP, designated MFRP174delG, which results in a complete knockout at the protein level. MFRP is a transmembrane protein expressed specifically in the RPE and ciliary body epithelium. In humans, mutations in MFRP cause severe nanophthalmos. In some cases, retinal degeneration has been noted as well. We have shown definitively in Mfrp174delG mice that the white fundus flecks are F4/80+ inflammatory cells, and are investigating whether a misregulated immune response is actively contributing to the pathology.
Methods: Complement factors in the retina were analyzed by immunocytochemistry. The origin of subretinal inflammatory cells was determined by lethal irradiation of mutant animals followed by bone marrow transplant using YFP+ mfrp+/+ donors.
Results: We observed decreased levels of C3d in mutant RPE. Mutants receiving bone marrow transplants from mfrp+/+ donors did not exhibit changes in disease progression, and subretinal cells were clearly donor-derived.
Conclusion: Immunolabeling experiments suggest that the alternative complement pathway may be misregulated. Transplant experiments suggest that the subretinal cells are derived directly from bone marrow precursors and that they are not primarily responsible for the retinal degeneration. We believe that mfrp173delG mice are a useful model for advanced non-exudative age-related macular degeneration.
Back to top
Identification of ganglion cell fate determinants through single cell transcriptomics
Jillian Goetz, Jeff Trimarchi
Department of Genetics, Development and Cell Biology, Iowa State University
Purpose: Retinal ganglion cells comprise such a small portion of the total retinal population that isolation of their individual transcriptomic signatures is impossible through analysis of the entire retina. To comprehensively discern the critical sets of genes involved in the cell fate decision process and in the early steps of ganglion cell development and diversification we are comparing the gene expression signatures from individual developing retinal ganglion cells isolated from the mouse, chicken and zebrafish.
Methods: Single cells were isolated from dissociated retinas at different developmental stages in the mouse, chicken and zebrafish. A PCR-based assay was used to determine the Math5 and Cath5 expressing cells, while the zebrafish cells were isolated from an Ath5:GFP transgenic zebrafish. The cDNAs resulting from these individual retinal cells were hybridized to Affymetrix arrays.
Results: Gene clusters were identified that correlate specifically with cells transitioning to the ganglion cell fate. These clusters are comprised of many different genes, including several transcription factors whose function in the developing retina is as yet unknown. In addition to the insight into ganglion cell development that these clusters have provided, they have also identified a host of marker genes for newborn ganglion cells.
Conclusions: Comparative analyses of the transcriptomes of developing retinal ganglion cells has allowed for the isolation of gene networks correlated among different species. These experiments enable us to focus our future functional experiments on those networks that are the most conserved and, therefore, the most likely to be critical in ganglion cell development.
Back to top
Optical Coherence Tomography (OCT) and histologic examination of a mouse model of chorioretinal coloboma
D. Gratie, S. Bhattarai, M. Stunkel, E. Schindler, M. Anderson, R.F. Mullins, AV. Drack
Department of Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, IA
Purpose: To characterize a chorioretinal coloboma occurring in the JF1 mouse line.
Methods: Indirect ophthalmoscopy with a 90D lens was performed on JF1 mice. Mice with chorioretinal colobomas had either optical coherence tomography (OCT) performed, followed by sacrifice and histologic preparation of the eyes, or were bred. Select mice fundi were photographed. Offspring of affected mice were examined for presence or absence of coloboma by indirect ophthalmoscopy.
Results: 60 eyes of 30 JF1 mice were examined. 19 were female, 11 were male. 11 of 30 mice demonstrated chorioretinal (CR) coloboma. 10/11 were unilateral. 6 were right eye, while 4 were left eye. 6 of 11 affected mice were female. All colobomas were adjacent to the optic nerve but none involved the nerve itself. On both OCT and histologic examination, the retinal layers adjacent to the colobomatous defect appeared to deviate course toward the sclera. The retinal pigment epithelium (RPE) ended abruptly on either side of the retinal defect. Rather than bare sclera, the base of the coloboma has a layer of collagen above sclera. Trichrome stain revealed collagen infiltrate into the retina. Based on pedigrees, this CR coloboma may be inherited in an autosomal dominant or autosomal recessive manner with variable expressivity.
Conclusions: We describe a mouse model of chorioretinal coloboma. The retinal defect appears to be complex, with disruption of normal retinal layers, and replacement of some tissue rather than simply a localized deficiency of retinal tissue. Variable expressivity is the norm, similar to human CR colobomas.
Back to top
Identification and Characterization of Genetic Factors Responsible for Cavitary Optic Disk Anomalies
Ralph J. Hazlewood1, Benjamin R. Roos1, Robert A. Hankonen2, Lee M. Jampol3, Wallace L.M. Alward1, John H. Fingert1
1Department of Ophthalmology and Visual Sciences, Carver College of Medicine, University of Iowa, Iowa City, IA; 2Department of Ophthalmology, State University of New York at Stony Brook, Stony Brook, NY; 3Department of Ophthalmology, Feinberg School of Medicine, Northwestern University, Chicago, IL
Purpose: Glaucoma is an acquired optic neuropathy that presents with visual field loss and progressive degeneration of the optic nerve resulting in retinal ganglion cell death. One way to study glaucoma is to investigate similar forms of optic nerve disease. Patients with cavitary optic disc anomalies (CODA) have congenital excavation of the optic nerve that, in some, progressively deteriorate in a manner similar to that observed in normal tension glaucoma patients. Consequently, we have studied CODA as a model of the optic nerve disease in glaucoma by searching for the gene that causes autosomal dominant inheritance of CODA in a large family.
Methods: Prior linkage studies mapped the CODA gene to a 13.5Mb segment of chromosome 12q14 (maximum non-parametric linkage score = 21.7). We have examined the linked region for the gene that causes CODA using comparative genome hybridization (CGH) and expression constructs.
Results: Study of affected family members with CODA identified a copy number variation (CNV) within the previously linked locus. All family members with CODA had two extra copies of a 6kb segment of DNA and this CNV was not detected in unaffected family members or in normal controls.Subsequent reporter gene analysis revealed an increase in expression when the 6kb segment is cloned in HEK293T cells.
Conclusions: We report a CNV within the previously linked region that is co-inherited with CODA in our family. We hypothesize that this CNV leads to dysregulation of gene expression and ultimately to the development of CODA. Ongoing research includes generating animal models.
Back to top
Genetic Modifiers of Secondary Glaucoma
Adam Hedberg-Buenz1, Michael G. Anderson 1,2
Departments of 1Molecular Physiology and Biophysics, 2 Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, IA
Purpose: Exfoliation syndrome and pigment dispersion syndrome are important glaucoma risk factors with strong hereditary components. C57BL/6J mice with the Lystbg-J and DctSlt-lt3Jmutations recapitulate aspects of exfoliation syndrome and pigment dispersion syndrome, respectively. The purpose of the current experiments was to determine the extent to which phenotypes of these mice are sensitive to genetic background by generating and analyzing congenic strains of mice in which these mutations have been bred onto the DBA/2J background.
Methods: Inbred C57BL/6J strains carrying either the Lystbg-J or DctSlt-lt3Jmutations were reiteratively backcrossed to DBA/2J mice for 10 generations. Anterior chamber phenotypes were assessed using slit lamp photography. Iris transillumination defects (iris-TID) were quantified and averaged among mice of identical genotype, with experimental and control cohorts tested for potential differences using Student’s t-test. Eyes and optic nerves were also compared histologically. All analyses were performed using age-matched DBA/2J controls.
Results: Lystbg-J and DctSlt-lt3Jiris phenotypes were both highly sensitive to genetic background. For Lystbg-J, compared to C57BL/6J, the DBA/2J genetic background resulted in a significant enhancement of the “Marcel-like” iris-TIDcommon in exfoliation syndrome. Quantification of iris transilluminancerevealed a 4-fold increase in defect severity in Lystbg-J irides on the DBA/2J compared to C57BL/6J background (n=8 eyes each strain). For DctSlt-lt3J, the DBA/2J genetic background caused a striking enhancement of iris atrophy and pigment dispersion. Interestingly, histologic analyses demonstrated that the DctSlt-lt3J mutation influenced the type and size of cellular infiltrate in the anterior chambers of DBA/2J mice. Surprisingly, neither of the DBA/2J congenic strains developed optic nerve degeneration.
Conclusions: These results indicate that the anterior chamber phenotypes associated with the Lystbg-J and DctSlt-lt3Jmutations are highly sensitive to genetic background. This finding may explain why genetic studies of pigment dispersion syndrome and exfoliation syndrome in human populations have been a challenge; genetic background is a significant confounding factor. Mechanistically, these results implicate a role for oxidative stress in pathophysiology of exfoliation syndrome and emphasize the importance of immune reactions in pathophysiology of pigment dispersion syndrome.
Back to top
Characterization of Synaptophysin’s Assembly and Subcellular Localization
Sarah R. Hengel, Sheila A. Baker, Yuan Pan, Modestos A. Modestou, Joe G. Laird
Department of Biochemistry, The University of Iowa
Purpose: Synaptophysin (SYP) is the most abundant protein in synaptic vesicles. Mutations in SYP have been associated with X-linked mental retardation and schizophrenia. SYP is known to form oligomers and bind components of the vesicle fusion machinery. We biochemically characterized the native SYP complexes and tested if disulfide bond formation was necessary for trafficking to the synapse.
Methods: Blue Native Page was used to characterize the oligomerization state of SYP from bovine retina. An RFP-tagged SYP and a mutant of SYP in which all four cysteines which can participate in disulfide bond formation were mutated to serines (SYP-CΔS-RFP) were created. Transgenic tadpoles were created to assess the synaptic localization of these proteins. The MembraneMax system was used to produce purified SYP and SYP-CΔS mutant proteins in vitro.
Results: Under native conditions, two major forms (60 kD, 205 kD) of SYP are observed. The smaller form likely represents a dimer of SYP bound to its partner VAMP2. The larger would then be a multimer of this complex. Under reducing conditions the amount of the 205 kD form decreases. In order to analyze the oligomerization state of SYP-CΔS, we expressed and purified SYP and SYP-CΔS in vitro as nanolipoprotein particles. SYP-RFP and SYP-CDS-RFP both localized to the synapse when expressed in photoreceptors.
Conclusions: SYP in its native state exists in two major forms. The larger form is stabilized by disulfide bonds. However, mutants unable to form disulfide bonds are still capable of localizing to the synapse.
Back to top
Joubert Syndrome and nephronophthisis proteins ARL13B, PDE6D and CEP164 target INPP5E to the primary cilium
Melissa C. Humbert1,2, Katie Weihbrecht1,2,3, Charles Searby2,4, Yalan Li5, Robert M. Pope5, Val C. Sheffield2,4, Seongjin Seo1
Departments of 1Ophthalmology and Visual Sciences, 2Pediatrics, 3Genetics Program, The University of Iowa,4Howard Hughes Medical Institute, 5Proteomics Facility, The University of Iowa, Iowa City, IA
Purpose: Mutations affecting ciliary components cause a series of related genetic disorders in humans, such as nephronophthisis (NPHP), Joubert syndrome (JBTS), Meckel-Gruber syndrome (MKS), and Bardet-Bield syndrome (BBS), which are collectively termed ‘ciliopathies’. Recent studies have shown that ciliopathy related proteins form several functional networks that build and maintain the primary cilia. Our research is to characterize INPP5E, a prenylated protein associated with JBTS, by examining its subcellular localization, interacting partners, and the mechanism by which it is targeted to the cilia.
Methods: We performed serial deletion mutagenesis, immunofluorescence microscopy, siRNA mediated gene knock-down approaches, tandem affinity purification and co-immunoprecipitation analyses using both endogenous INPP5E (visualized by antibodies) and tagged INPP5E constructs stably transfected into HEK293T and IMCD3 cells.
Results: The ciliary targeting sequence of INPP5E is near its C-terminus and uses prenyl-binding-protein PDE6D dependent mechanisms. Ciliary targeting of INPP5E is facilitated by another JBTS protein, ARL13B, but not by ARL2 or ARL3. We further demonstrate that INPP5E interacts with several ciliary and centrosomal proteins, including a recently identified ciliopathy protein CEP164.
Conclusions: These findings suggest that ARL13B, INPP5E, PDE6D, and CEP164 form a novel functional network that is involved in JBTS and NPHP but is distinct from previously defined NPHP and MKS protein networks.
Back to top
Genetic complexity of the Central Corneal Thickness QTL 1 (Cctq1) locus
Demelza Koehn1, Maurisa Aimable1, Michael G. Anderson1,2
Departments of 1Molecular Physiology and Biophysics and 2Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, IA
Purpose: Thin central corneal thickness (CCT) is an important endophenotype of glaucoma. Our goal is to identify genes that regulate the magnitude of CCT, using it as an entry point for studying the etiology of glaucoma. Our previous work using a quantitative approach with intercrosses between mice with thin corneas (C57BLKS/J; KS) and thick corneas (SJL/J; SJL) identified Central corneal thickness QTL 1 (Cctq1), a ~15 cM region on mouse chromosome 7. Efforts to narrow this region using N4 congenic mice indicated the region was recalcitrant to division. This suggested either more than one CCT-regulating gene exists within the region or that there was too much genetic heterogeneity in the N4 mice. Here, we show evidence that an additional QTL exists adjacent to Cctq1, likely contributing to the genetic complexity of this trait.
Methods: To determine if more than one CCT-regulating gene exists within or near Cctq1, the original dataset was analyzed performing a focused pair-wise scan of chromosome 7 using R/qtl. Identified interacting loci were subsequently subjected to ANOVA in which one QTL was dropped at a time, which assesses support for the individual loci and their interactions.
Results: From the R/qtl analysis, we determined there is a second CCT-regulating QTL directly next to Cctq1 that has an epistatic interaction with Cctq1 (full LOD score = 10.1). Using the full model result and dropping one QTL at a time resulted in LOD scores of 8.4 (p=7.9(10)-7) and 7.0 (p=1.6(10)-5) for Cctq1 and the adjacent locus, respectively, indicating the interaction between these two loci is important for their effect.
Conclusions: These results suggest two adjacent QTL are located on mouse chromosome 7 which regulate CCT. We have generated N10 congenic mice harboring both QTL and continue to utilize these mice in a digenic mapping strategy designed to map the genes underlying these two QTL.
Back to top
Interaction between the immune and complement systems in glaucoma
Markus H. Kuehn
The University of Iowa, Department of Ophthalmology and Visual Sciences
Purpose: The degeneration of retinal ganglion cells (RGC) in the glaucomatous retina is associated with neuroinflammatory processes, including local synthesis and deposition of complement components 1q (C1q) and 3 (C3) on RGC. Data from several laboratories have indicated the presence of auto-antibodies directed against retinal epitopes in the serum of glaucoma patients, which could potentially result in the activation of the classical complement cascade. Here we evaluated whether C1q binding and subsequent activation of the complement cascade in glaucoma is dependent upon the presence of immunoglobulins using RAG-/- mice which do not develop mature T- and B- lymphocytes and do not produce immunoglobulins.
Methods: Experimental glaucoma was induced in C57BL/6J and B6.129S7-Rag1tm1Mom/J (RAG-/-) mice using a microbead occlusion approach. RGC densities as well as axonal damage were determined. The deposition of C1q and C3 was visualized using immunohistochemistry of retinal cryosections and confirmed by Western blot of retinal cell membrane protein preparations. Complement activation and accumulation were also evaluated in vitro using dissociated retinal cell cultures.
Results: Injection of microbeads into the anterior chamber of mice induces mild IOP elevation in the majority of treated eyes. Elevated intraocular pressure was maintained for up to 4 weeks. Complement component C1q can be observed in the retinal ganglion cell layer in both RAG-/- and control mice with elevated IOP. Our proteomic analysis indicated that C1q and C3 are membrane bound in both RAG-/- and control mice. The absence of immunoglobulins in RAG-/- mice did not affect the rate of axonal damage or RGC loss. In addition, RGC in retinal cultures maintained in serum-free media are also C1q and C3 immunoreactive, further suggesting that Ig is not required for C1q and C3 binding to damaged RGC.
Conclusion: Our data demonstrate that lack of immunoglobulins and mature T/B cells does not influence the progression of glaucoma. Furthermore, immunoglobulins do not appear to be required for C1q binding and complement cascade activation on damaged RGC. These findings suggest that C1q recognizes an alternative binding partner expressed by stressed RGC.
Back to top
Analysis of proliferation and cellular differentiation in a zebrafish model for retinoblastoma
Deborah E. Lincow1, Pavel A. Brodskiy2, Jeffrey J. Essner3, Donald S. Sakaguchi3,4, and Maura McGrail3
1Santa Monica College, Santa Monica, CA; 2Department of Chemical and Biological Engineering; 3Department of Genetics, Development, and Cell Biology; 4Neuroscience Program, Iowa State University, Ames, IA
Purpose: Retinoblastoma, the third most common childhood cancer, is a developing cancer resulting in malignancy of retinal cell layers. The purpose of this study was to characterize the pattern of proliferation of retinoblastoma in a mutant strain of zebrafish.
Methods: Genetic retinal tumors are expressed in Tol2 transposon transgenic line Tg(flk1:RFP)is18 zebrafish. We examined retinal cell proliferation in 4.5 month-old Tg(flk1:RFP)is18 zebrafish, and their non-transgenic siblings, treated with 5 mM bromodeoxyuridine (BrdU). Immunostaining for BrdU along with antibodies specific for different retinal cells was conducted.
Results: 81% of eyes examined showed abnormal development. This included hyperplasia, gliosis, and loss of laminar structure. Seven of the mutant eyes had ectopic cells outside of the normal ganglion cell layer (GCL), and expressed glial fibrillary acidic protein (GFAP), BrdU, or both. SOX2, a stem cell marker, was expressed in many cells, including cells expressing BrdU. Brain lipid-binding protein (BLBP), a marker for immature Müller glia, was expressed in many of the mature retinas. In tissues treated with glutamine synthetase (GS) and BLBP, cells were labeled within close proximity. Large masses of GFAP+ cells were found in the ganglion cell layer.
Conclusions: 4.5 month-old Tg(flk1:RFP)is18 zebrafish have a high frequency of ocular tumors. Coexpression of SOX2 and BrdU supports a progenitor cell type of origin. Understanding the mechanism for how retinal tumors are formed in zebrafish may provide important insight into the progression of this disease in humans.
Back to top
Localization of Cav1.4 Ca2+ channels in photoreceptor synapses in the adult and developing retina
X. Liu1, V. Kerov2, S. Baker2, S. H. DeVries3, F. Haeseleer4, A. Lee1
1Mol. Physiol. & Biophysics, 2Biochem., Univ. of Iowa, Iowa City, IA; 3Opthalmology, Northwestern Univ., Chicago, IL; 4Physiol. & Biophysics, Univ. of Washington, Seattle, WA
Purpose: Cav1.4 channels mediate L-type Ca2+ currents that trigger tonic release of glutamate from photoreceptor synaptic terminals. To gain insights into the function of Cav1.4 during development, we analyzed the localization of Cav1.4 at rod and cone synapses at various postnatal ages.
Methods: We generated antibodies that recognize Cav1.4 and double-labeled with antibodies against a presynaptic protein, RIBEYE.
Results: Cav1.4 was strongly colocalized at rod synapses in mice and cone synapses in ground squirrels. Cav1.4 immunofluorescence was specific in that it was observed in synapses in the outer plexiform layer (OPL) of wild-type but not Cav1.4 knockout (KO) mice. In Cav1.4 KO mice, photoreceptor synapses were abnormally spherical in shape. During development, Cav1.4 antibodies first labeled puncta at P5 when RIBEYE staining starts to concentrate in the newly forming OPL. From P6 to P10, the intensity of Cav1.4 labeling in the OPL increases, but does not completely colocalize with RIBEYE-labeling. Cav1.4/RIBEYE-positive structures are spherical and become larger and more numerous with postnatal age. At P11, both Cav1.4/RIBEYE-positive structures first become “horseshoe”-shaped with strong colocalization of the two signals. Starting from P13, the age of eye opening in mice, Cav1.4 and RIBEYE completely colocalize at synapses with the characteristic mature morphology in adult mice.
Conclusions: At early stages, Cav1.4 and RIBEYE can form independent clusters prior to OPL formation and Cav1.4 is necessary for normal synaptic ribbon morphology. Our data provide additional evidence that presynaptic Cav1.4 channels are positioned to regulate transmitter release in both mammalian rods and cones.
Back to top
Onecut transcription factors play distinct roles in the developing and mature retina
Gregory M. Martin, Jeffrey Trimarchi
Department of Genetics, Development and Cell Biology, Iowa State University
Purpose: To gain insight into the gene networks that drive retinal ganglion cell fate, we determined the transcriptomes of individual Math5-expressing progenitor cells. We found significant clusters of genes that correlated specifically with distinct subsets of Math5+ cells. These clusters contained many different transcription factors whose function in the developing retina was unexplored. Here we report on an analysis of retinal development in mice individually deficient for two of these transcription factors, Onecut1 and Onecut2.
Methods: We examined the retinas of mice deficient for either Onecut1 or Onecut2 by three main methods. We compared wildtype and knockout retinas by microarray analysis, in situ hybridization and immunohistochemistry. We used a variety of probes and antibodies to examine each of the different retinal cell types in these knockout mice. These analyses were performed at multiple developmental time points and in the adult.
Results: The combination of in situ hybridizations and immunostainings of the Onecut2 deficient adult retinas showed that all of the retinal cell types are present. Interestingly, however, horizontal cells are reduced in the adult Onecut2 retina and the developing Onecut1 retina. Microarray analysis of these retinas showed a consistent decrease in Lhx1, providing a potential explanation for the horizontal cell loss.
Conclusions: We found the Onecut transcription factors to be expressed in Math5+ progenitor cells. Analysis of mice deficient for either Onecut1 or Onecut2 revealed a critical role for these transcription factors in the generation of a large percentage of horizontal cells.
Back to top
Mutant Human Myocilin Induces Strain Specific Differences in Ocular Hypertension and Optic Nerve Damage in Mice
Colleen M. McDowell1, J. Cameron Millar2, Simon W.M. John3, Iok-Hou Pang2, Abbot F. Clark1
1North Texas Eye Research Institute, Department of Cell Biology and Anatomy, University of North Texas Health Science Center, 2Alcon Research, Ltd., Fort Worth, TX, 3Howard Hughes Medical Institute, The Jackson Laboratory, Bar Harbor, ME
Purpose: Elevated intraocular pressure (IOP) is a causative risk factor for the development and progression of glaucoma. Glaucomatous mutations in myocilin (MYOC) damage the trabecular meshwork and elevate IOP in humans and mice. The goal of this work is to develop the first inducible mouse model of POAG using a human POAG relevant transgene (i.e. mutant MYOC) expression in mouse eyes to elevate IOP and cause pressure induced damage to the optic nerve.
Methods: Four mouse strains (A/J, BALB/cJ, C57BL/6J, and C3H/HeJ) were used in this study. Ad5.MYOC.Y437H (5 X 107 pfu) was injected intravitreally into one eye, with the contralateral eye serving as the control eye. Conscious IOP measurements were taken using a rebound tonometer. Optic nerve damage was determined by scoring PPD stained cross sections. Retinal ganglion cell and superior colliculus damage was assessed by Nissl stain cell counts.
Results: Ad5.MYOC.Y437H caused a statistically significant IOP elevation in BALB/cJ, A/J, and C57BL/6J mice. IOPs increased to approximately 25 mm Hg for 8 weeks (p<0.0001). The C3H/HeJ mouse strain was resistant to Ad5.MYOC.Y437H induced IOP elevation. Only the A/J strain had considerable and significant optic nerve damage at the end of 8 weeks with optic nerve damage score of 2.64 +/- 0.19 (n=18, p<0.001) in the injected eye. There was no statistical difference in retinal ganglion cell death or superior colliculus damage at the 8-week time point in any of the strains tested.
Conclusion: These results demonstrate strain dependent responses to Ad5.MYOC.Y437H-induced ocular hypertension and pressure-induced optic nerve damage.
Back to top
Sensitivity of the nee mutation to genetic background
Kacie J. Meyer1, Michael G. Anderson 1,2
Departments of 1Molecular Physiology and Biophysics, 2 Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, IA
Purpose: The nee mouse strain, harboring a recessive mutation in Sh3pxd2b, exhibits an early onset and severe developmental glaucoma with elevated intraocular pressure (IOP) followed by optic nerve head excavation and axon loss. SH3PXD2B is required to produce normally functioning podosomes, which are cellular protrusions of the plasma membrane that bind and degrade extracellular matrix. Because the nee mutation spontaneously arose on an infrequently used B10 genetic background, the extent to which genetic background influences nee phenotypes was unclear.
Methods: To ascertain the effect of genetic background on ocular phenotypes of the nee allele, we first produced an N10 congenic strain transferring the Sh3pxd2bnee mutation onto a C57BL/6J genetic background. Subsequently, we initiated an intercross between B6-Sh3pxd2bnee and CAST/EiJ, a wild-derived inbred mouse strain. Ocular phenotypes of nee mice from both experiments were compared using slit-lamp exams, optical coherence tomography imaging, and IOP measurements.
Results: Indistinguishable from the B10-nee mice, homozygosity of the nee mutation on the B6 background resulted in peripheral anterior synechia upon examination at P17 as well as cloudy corneas and enlarged anterior chambers by 3 months of age. In contrast, the ocular phenotypes of F2 progeny from the intercross between B6-Sh3pxd2bnee and CAST/EiJ display substantial variable expressivity and many mice have no apparent synechia.
Conclusions: The generation of the B6-Sh3pxd2bnee congenic mouse, which recapitulates the phenotypes published using the B10 background, will be a valuable resource for future studies of glaucoma. In addition, the overt and tractable ocular phenotypes of B6-Sh3pxd2bnee mice, which are suppressed in the F2 mixed genetic background, provide the opportunity to identify modifier genes contributing to pathological molecular pathways for glaucoma. Future studies will focus on mapping the gene(s) responsible for the phenotypic suppression reported here and understanding the role of Sh3pxd2b and podosomes in adult mice.
Back to top
Characterization of HCN1 trafficking in rod photoreceptors
Y. Pan, J.G. Laird, S.A. Baker
Department of Biochemistry, The University of Iowa, Iowa City, IA
Purpose: Hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) is a non-selective cation channel that is expressed in retina, brain and heart. It functions by shaping membrane potential and synaptic output. In photoreceptors, HCN1 is primarily localized in the membrane of inner segments and to a lesser extent in synaptic termini. TRIP8b, an accessory protein of HCN, regulates channel activity and localization in the dendrites of cortical and hippocampal neurons. However, it is not known how HCN1 is targeted to the inner segments of photoreceptors or if TRIP8b plays a role in this process. We studied HCN1 trafficking in photoreceptors.
Methods: Various regions of HCN1 were fused to a membrane reporter and expressed in transgenic Xenopus rods. Localization of the transgenically expressed proteins was visualized with confocal microscopy.
Results: We found that a region in the C-terminus of HCN1 contains a strong synaptic targeting signal. This signal has been mapped to amino acids 726-887 of human HCN1. Interestingly, the synaptic targeting signal does not contain the binding sites for TRIP8b.
Conclusion: We have identified a novel region within the C-terminus of HCN1 that is sufficient for targeting to synaptic termini in rods. However, the endogenous HCN1 localizes predominantly to the rod inner segments. This indicates that the synaptic targeting of HCN1 is down-regulated in rods, perhaps by TRIP8b interaction through other regions of HCN1.
Back to top
Voted Outstanding Oral Presentation
Overexpression of the cilia protein RPGRIP1L rescues rhodopsin expression in cux1b morphant zebrafish
Pamela R. Pretorius1, Julia M. Hatler1,3, Stephanie L. Lerach1, Ashley M. Spahn1, Elizabeth Speltz3, Lisa A. Schimmenti1,2,3
Departments of 1Pediatrics, 2Department of Ophthalmology and Visual Neurosciences, and 3Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN
Purpose: Proper eye development during embryogenesis is critical for vision function. Several transcription factors are required for normal ocular morphogenesis, visual cell differentiation and maintenance of cell function. Studies in Drosophilia identified sparkling (spa) as an ortholog to the human transcription factor PAX2. Spa mutants have abnormal eye development and reduced expression of the transcription factor cutl1/cux1b in cone cells. Cux1b regulates expression of the ciliary gene Rpgrip1l, which coordinates leptin receptor trafficking to the cilia. We hypothesized that cux1b has a role in vertebrate retinal development and photopigment trafficking.
Methods: The genetic and functional similarities to mammalian eyes make zebrafish an ideal model to study vertebrate eye development. An antisense oligonucleotide Morpholino was utilized to knockdown cux1b gene expression. Immunohistochemistry was used to assess cux1b morphant and rescued phenotypes in the retina.
Results: Microinjection of a morpholino against cux1b into zebrafish embryos results in microphthalmia. Histological and immunohistochemical analysis reveal normal cone and rod photoreceptor cell specification in cux1b knockdown embryos; however, outer segments fail to fully develop and have reduced rhodopsin expression. Moreover, green opsin expression was not restricted to the photoreceptor outer segment, but was also present in the cell body. Overexpression of RPGRIP1L mRNA in cux1b knockdown embryos normalizes eye size and partially restores rhodopsin expression.
Conclusions: These data provide evidence that cux1b is required for retina development and maintenance of photoreceptor outer segments. Given the rescue of rhodopsin expression following RPGRIP1L overexpression, we propose that cux1b transcriptionally regulates rpgrip1l expression in the retina.
Back to top
Molecular mapping of the zebrafish retinoblastoma-like tumor model Tg(flk1:RFP)is18
Staci L. Solin1, Ying Wang1, Pavel A. Brodskiy1, Deborah Lincow1, Elizabeth M. Whitley2, Jeffrey J. Essner1, Donald S. Sakaguchi1, Maura McGrail1.
1Genetics, Development and Cell Biology; 2Veterinary Pathology; Iowa State University, Ames, IA, 50011. email: mmcgrail@iastate.edu.
We have isolated a novel retinal tumor line in zebrafish that models human inherited retinoblastoma. In the zebrafish transgenic line Tg(flk1:RFP)is18 ocular tumors develop in heterozygous adults as early as 5 months of age. We previously demonstrated that the retinal tumors show cytological and histological similarities to human retinoblastoma. The zebrafish retinoblastoma-like locus is genetically linked to the Tg(flk1:RFP)is18 transgene integration site and has been transmitted through 6 generations.
Purpose: To identify the gene disrupted at the transgene integration site in the Tg(flk1:RFP)is18 retinoblastoma-like model.
Methods: A custom SureSelect Target Enrichment kit was designed to capture the transgene and flanking genomic DNA from line Tg(flk1:RFP)is18. Captured sequences were paired-end sequenced on an Illumina GAIIX and mapped to the zebrafish V9 genome.
Results: Five Tg(flk1:RFP)is18 samples contained paired-end sequences that mapped to the transgene and to a unique location at position 24.2 Mb on chromosome 3. PCR and genomic southern blot analyses confirm the Tg(flk1:RFP)is18 integration site is located in the second intron of the macro non-coding RNA cbx1aOppositeStrandTranscript, cbx1aOST. RT-PCR demonstrates thatcbx1aOST is expressed in wild type adult retina.
Conclusions: The results suggest that the molecular lesion in the Tg(flk1:RFP)is18 line disrupts expression of the macro non-coding RNA cbx1aOST. We are investigating whether expression of cbx1aOST is altered in Tg(flk1:RFP)is18 and tumor tissue. Macro non-coding RNAs are implicated in epigenetic mechanisms controlling gene expression. Future studies will test the role of cbx1aOST in epigenetic mechanisms that function in retinal stem cell proliferation, cell fate specification, and tumorigenesis.
Back to top
Electrophysiological Changes in Animal Models of Leber’s Congenital Amaurosis
Kelsey N. Spalding, Steven F. Stasheff, Michael P. Andrews, Frederick R. Blodi, Malani Shankar
Purpose: Leber’s Congenital Amaurosis is a hereditary eye disease causing rapid visual deterioration in children. We hypothesize that early retinal degeneration disrupts the formation of inner retinal circuits, distorting visual signals such that they are no longer recognized by the brain. Thus, an effective photoreceptor-restoring treatment must be instituted early enough to prevent the disruption of downstream retinal circuits. Until now, few studies have described the electrophysiology of two mutations of particular interest: Rpe65, the only form currently treatable with gene therapy, and Cep290, the most common form of the disease.
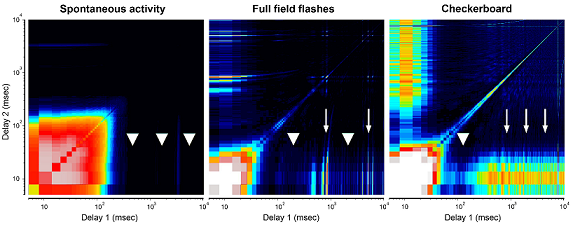
Methods: Using in-vitro multielectrode action potential recordings, we studied the spontaneous and light evoked responses of retinal ganglion cells in Rpe65, Cep290, and wild-type mice prior to eye-opening through adulthood.
Results: Before eye-opening, normal developmental waves of correlated firing appear in both Rpe65 and Cep290. In both strains, hyperactivity emerges shortly after eye opening. Rpe65 retinal ganglion cells have virtually no light response at any age, while Cep290 cells have reduced responses at early ages that continue to decay at a slower rate than in other degenerative lines. Additional features that distinguish the strains include the spectrum of spontaneous firing frequencies, the shape of intensity-response curves, and the on/off response ratios.
Conclusions: The time courses of changes in the electrophysiological characteristics of Rpe65 and Cep290 retinal ganglion cells identified in this study could provide key insight into the potential success of treatments such as gene therapy, stem cell transplantation, or electrical stimulation, and the optimal time of treatment administration.